CAPÍTULO 7 OJO SECO ASOCIADO A DERMOPATÍAS Andrea Sanz López |
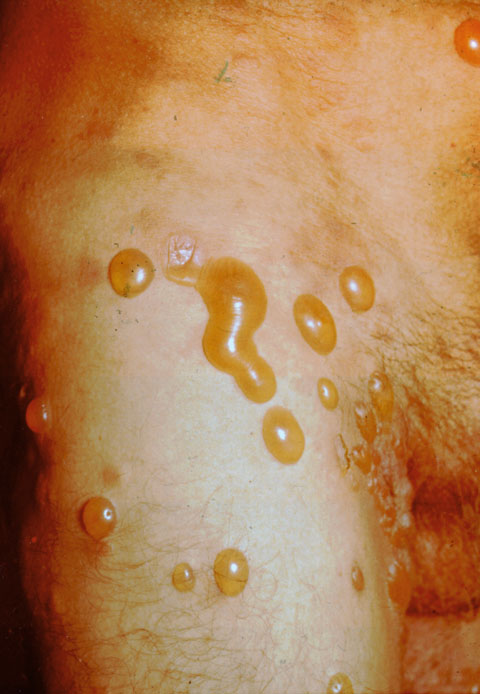
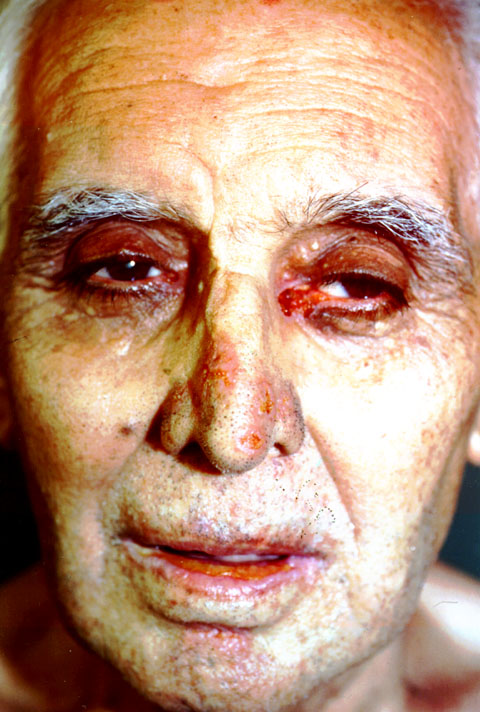
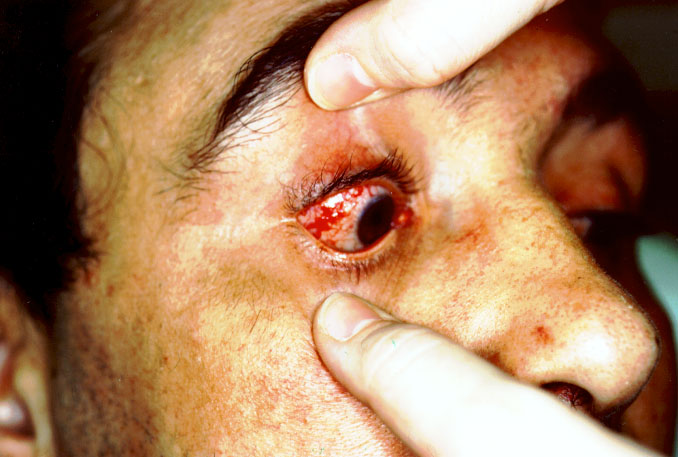
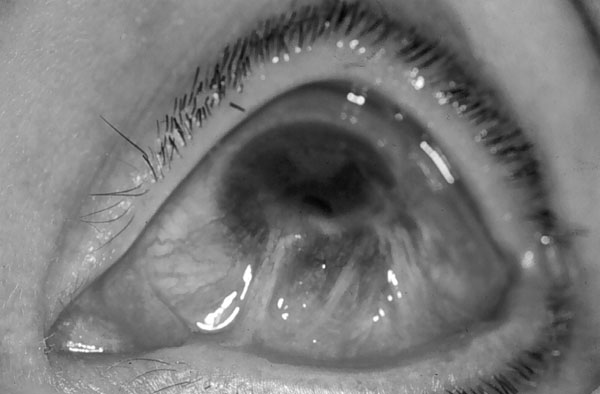
| PÉNFIGO VULGAR El pénfigo es una enfermedad crónica, en la que se forman ampollas intradérmicas por acantolisis. La enfermedad suele aparecer en personas de mediana edad. En el 50% de los pacientes las lesiones aparecen en las mucosas antes de hacerlo en la piel. La mucosa más afectada es la oral, pero también pueden aparecer lesiones en conjuntiva ocular, esófago, vulva y recto. A los pocos meses de iniciarse las lesiones orales, comienza la fase bullosa generalizada (figuras 7-1 y 7-2). Estas lesiones son dolorosas, de lenta curación y no dejan cicatriz. Cuando afecta al ojo lo hace generalmente a la parte cutánea de los párpados (figura 7-3) y más raramente, a la conjuntiva palpebral y bulbar (figura 7-4).
El pénfigo puede estar facilitado por factores ambientales. Así, se han descrito variedades circunstanciales y reiteradas. Por ejemplo en Brasil se ha descrito el pénfigo brasileño (fogo selvagem) o síndrome de Améndola (Améndola 1945, Korting 1973) , endémico en todos los grupos raciales del estado de Sao Paulo, que afecta casi constantemente la piel de los ojos y frecuentemente la conjuntiva, y que en un 5% de los casos produce opacidades cristalinianas.
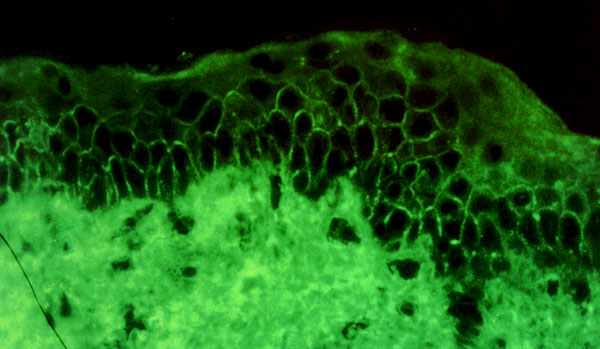
El diagnóstico del pénfigo se hace por la clínica y por la presencia de IgG, IgM e IgA y complemento en la sustancia intercelular de la piel y mucosas afectadas (figura 7-5). El signo de Nikolsky (despegamiento de la piel sana que rodea una ampolla cuando se presiona con el dedo) es positivo.
El tratamiento de elección son los corticoides asociados en los casos graves a azatioprina o metotrexate.
PENFIGOIDE Histológicamente los penfigoides se diferencian del pénfigo en que en los penfigoides la lesión principal es la destrucción de la membrana basal del epitelio mucocutáneo por presencia de anticuerpos anti-membrana basal, dando por tanto ampollas subepiteliales, mientras que en el pénfigo la lesión básica es la acantolisis (destrucción de los puentes intercelulares), dando por tanto ampollas intraepiteliales. El penfigoide suele aparecer en personas mayores (de 65 a 75 años), manifestándose como una urticaria generalizada con lesiones eritematosas que evolucionan a ampollas subepidérmicas. Estas ampollas son casi indoloras, perduran largo tiempo, y si se rompen curan rápidamente. La participación de las mucosas no es frecuente, siendo la más afectada la oral, y más raramente la conjuntival. Se pueden demostrar anticuerpos anti-membrana basal epidérmica. Además, en la dermis aparece un infiltrado inflamatorio con eosinófilos, neutrófilos, linfocitos e histiocitos. El tratamiento de elección son los corticoides.
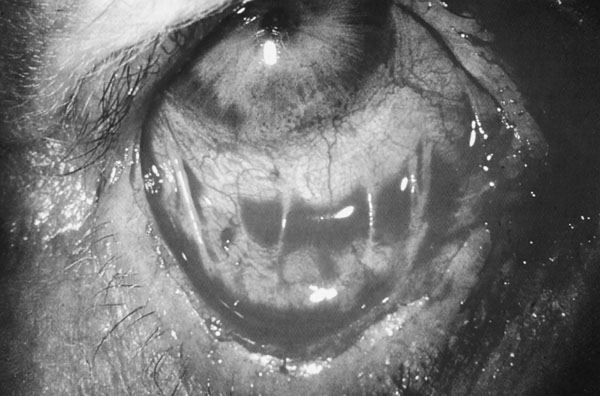
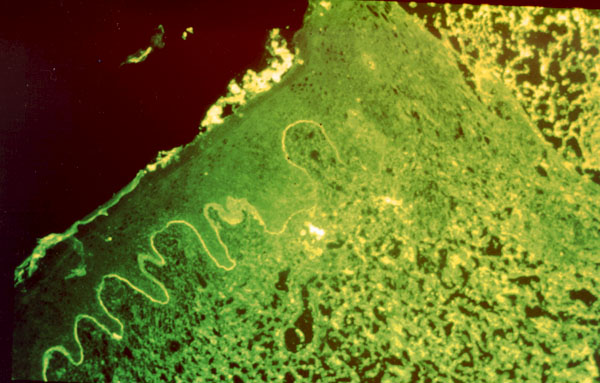
PENFIGOIDE OCULAR CICATRICIAL El penfigoide ocular cicatricial, también conocido como penfigoide mucoso benigno, dermatitis bullosa mucosinequiante y atrofiante o penfigoide de Lortat Jacob, es un tipo de penfigoide que afecta principalmente a la conjuntivas. Es el de 1/15.000 al 1/40.000 de los enfermos de una consulta oftalmológica (Díaz Valle et al 1993). Afecta con más frecuencia a mujeres de edad media o avanzada. Suele iniciarse y afectar principalmente a la conjuntiva, pero también puede manifestarse en otras mucosas y, más raramente, en la piel de zonas periorificiales. Clínicamente aparecen ampollas subepidérmicas, no dolorosas, con gran tendencia a la cicatrización. El comienzo, generalmente, es unilateral, pero en los dos años siguientes siempre se bilateraliza. Se inicia como una conjuntivitis crónica progresiva. La enfermedad cursa crónicamente, con períodos alternantes de actividad y remisión. Las ampollas conjuntivales, a menudo fugaces por romper por el frote de los párpados, dejan cicatrices que se siguen de hiposecreción lacrimal por destrucción de la conjuntiva y oclusión de los conductos de las glándulas lacrimales. Además puede aparecer simbléfaron, entropion y triquiasis, y en muchos casos se llega a la opacificación total de la córnea. Las complicaciones oculares son las únicas importantes. Según Balestrazzi et al 1979 el primer signo clínico es la desaparición de las células caliciformes. Después sobreviene una transformación fibrosa de la mucosa, y ésta se atrofia y se retrae. En las fases iniciales puede haber una xerosis mucindeficiente con secreción acuoserosa cuantitativamente normal (Norn 1972) o que haya una relación de componentes acuoseroso/mucínico alta (Carroll et al 1968, Dohlman et al 1976). Si la enfermedad progresa, se retrae la mucosa conjuntival (figura 7-6), se obliteran los dacriodocos y se agrava la deficiencia acuoserosa, hasta poder haber un ojo seco absoluto.
La clasificación de Foster 1986 es como sigue: Estadío I: Cicatrización subconjuntival
y fibrosis Aparecen depósitos de IgG y C3 en la membrana basal de las mucosas, e invasión del epitelio mucoso afectado por lifocitos, histiocitos y algunos eosinófilos (figura 7-7). Posteriormente aparecen fibloblastos que darán lugar a la fibrosis cicatricial característica que da nombre a la enfermedad.
La enfermedad responde mal al tratamiento, siendo el más usado el tratamiento sistémico con corticoides, ciclofosfamida, azatioprin, y/o dapsona y, actualmente, la ciclosporina A. En cuanto al tratamiento tópico, se prescriben lágrimas artificiales y antiinflamatorios.Bisantis 1976 mejoró grandemente dos casos con instilaciones de un coliro de autosuero diluido, repetido varias veces diarias durante varios meses. La oclusión de los puntos lacrimales puede ser beneficiosa en los estadios iniciales; posteriormente suelen obliterarse ellos espontáneamente.
EPIDERMOLYSIS BULLOSA ACQUISITA Enfermedad bullosa autoinmune que puede confundirse con el penfigoide ocular cicatricial, pues ambas provocan fibrosis de la conjuntiva. Si se hace biopsia, se ven bajo la lámina densa y entre las fibrillas de anclaje depósitos de inmunocomplejos.
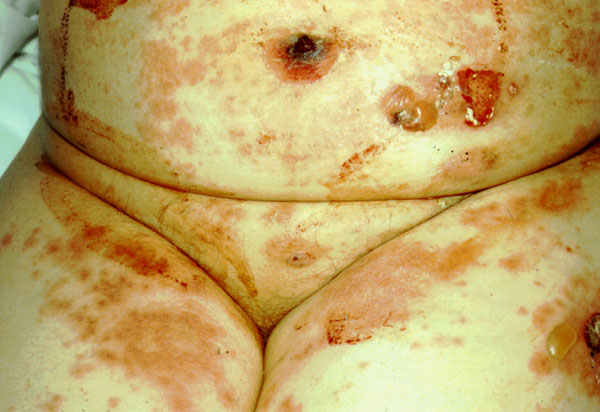
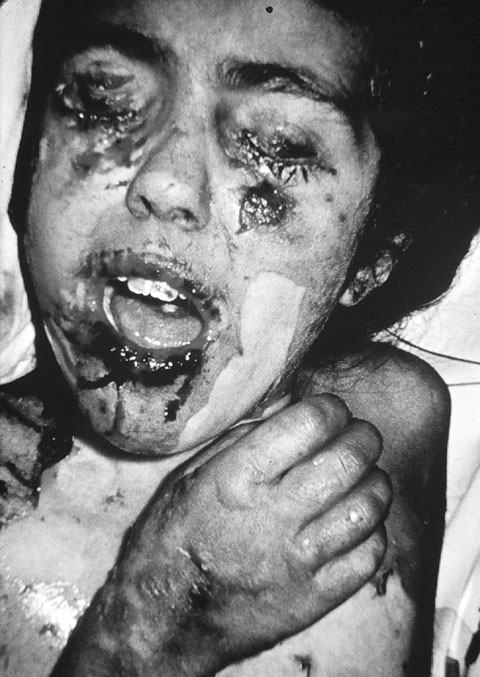
SÍNDROME DE STEVENS-JOHNSON El síndrome de Stevens-Johnson es un eritema multiforme severo, de comienzo brusco, que se sigue de la formación de ampollas subepidérmicas. Suele aparecer horas, días o semanas tras el inicio de la recepción de un fármaco. En el primer caso, se supone que el paciente estaba previamente sensibilizado al medicamento, y en el segundo, que se sensibiliza durante la toma continuada del medicamento. Los casos más frecuentes se dan por inhibidores de la anhidrasa carbónica como acetazolamida (Nayak et al 1981) y metazolamida (Shirato et al 1977). Parece haber una predisposición genética, pues los casos por metazolamida se dan casi siempre en japoneses y coreanos con haplotipo HLA-B59, específico de estas razas. El cuadro clínico empieza por fiebre ligera y aparición de hinchazón de piel y mucosas y de un eritema multiforme con sus típicas lesiones en diana (centro eritematoso rodeado de una anillo blanco, rodeado a su vez de un halo eritematoso). Las lesiones progresan rápidamente a bullas extendidas por todo el cuerpo, que, pese a un intenso tratamiento inmunosupresor, acaban rompiendo (figura 7-8).
En los ojos puede aparecer eritema, ampollas, y erosiones epiteliales corneales determinantes de disminución de visión, lo que se acompaña de hiperlacrimación. Las lesiones evolucionan en unos meses a la desecación y descamación. El tisc/but baja (Lemp et al 1970b, Baum 1973, Dohlman et al 1976). Pese al bajo tisc/but, la lágrima de estos pacientes suele tener una baja tensión superficial, incluso inferior a lo normal, protegiéndolos un poco de la desecación normal esta baja tensión superficial parece deberse a la presencia en lágrima de productos inflamatorios y no a un incremento de la mucina, pues ésta aparece disminuida (Dohlman et al 1976, Holly et al 1977b). En los casos graves quedan retracción de los pliegues lacunares y de los fórnices, simbléfaron y leucomas vascularizados (figura 7-9).
El diagnóstico se hace por la clínica, pues las pruebas epicutáneas e intradérmicas al fármaco causante suelen resultar negativas.
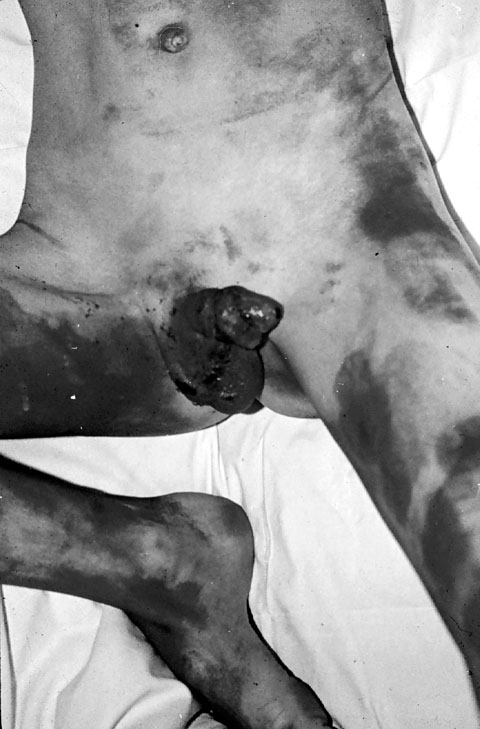
SÍNDROME DE LYELL Es una necrolisis epidérmica relacionada con la administración sistémica de medicamentos, especialmente de AINES, sulfamidas, hidantoinas, barbitúricos y penicilinas. También se ha sugerido a veces un desencadente infeccioso. Se inicia como un eritema perioral que se extiende en unas horas a todo el cuerpo y evoluciona hacia ampollas subepidérmicas (figura 7-10); las lesiones son dolorosas. El síndrome de Lyell suele ir acompañado de mal estado general, pirexia y anorexia. Generalmente cura en 15 días, dejando cicatriz. Sólo ocasionalmente se agrava, pudiendo llevar a la muerte.
Cuando afecta a la piel y mucosa de los ojos aparece conjuntivitis y dermatitis ampollosas, blefaritis, úlceras corneales, y finalmente entropion, triquiasis, simblefaron y leucomas corneales con KCS por destrucción mucosa y afectación acuoserosa directa, o por cierre de los dacriodocos. Las glándulas de Meibomio unas veces son respetadas (Marco et al 1974), pero otras participan en el proceso, incluso de manera preferente (Pesch et al 1967). Se trata con corticosteroides y con medidas para evitar la deshidratación y la infección mucocutánea.
SÍNDROME DE REITER Es un síndrome mucosinequiante, no ampolloso, que produce inflamación mucoide o purulenta de mucosas (aparato genital, boca y conjuntiva ocular) e inflamación de articulaciones. También puede aparecer uretritis, pleuritis, adenopatía periférica, fiebre, etc. La enfermedad suele aparecer en edades puberales o medias de la vida, y autolimitarse en unas semanas, dejando mayor o menor destrucción mucosa y articular. La afectación de la conjuntiva puede provocar una metaplasia escamosa, con disminución de células caliciformes y retracción conjuntival que va desde borramiento de los pliegues conjuntivales hasta grave simbléfaron. La córnea y la úvea pueden también afectarse. El origen del síndrome es desconocido. La frecuencia de HLA-B27 es muy superior a la de la población normal. Se piensa en una infección viral que modifique el sistema inmunológico. La búsqueda de virus y otros gérmenes en las mucosas afectadas, resulta negativa. La biopsia conjuntival muestra paraqueratosis epitelial y acantosis, con infiltración de linfocitos polimorfonucleares.
SÍNDROME DE ZINSSER-ENGMAN-COLE Este síndrome también conocido como Dyskeratosis congénita cum pigmentatione, es una atrofia de la dermis y epidermis, con queratinización de mucosas y depósitos pigmentarios en el corion mucocutáneo. En los ojos se manifiesta por conjuntivitis ampollosa y queratinización conjuntival, que evolucionan hacia retracción mucosa, ojo seco, grave opacificación corneal y retracción de los puntos lacrimales. En el resto del cuerpo se manifiesta por piel de "mármol" o "bronce", hiperpigmentación en difusa y en manchas, atrofias cutáneas y telangiectasias. El síndrome parece ser hereditario autosómico recesivo. Comienza a desarrollarse hacia la pubertad. |