|
INTRODUCCIÓN En este capítulo se pretende resumir parte de la experiencia que de forma ininterrupida durante más de 20 años ha conseguido la labor en equipo de varias generaciones de oftalmólogos en el Hospital Clínico San Carlos de Madrid, bajo la dirección del Prof. García Sánchez. En este Servicio de Oftalmología se consiguió reunir con los años una casuística de glaucomas congénitos y juveniles que, a pesar de la rareza de esta enfermedad, puede ser considerada una de las más importantes a nivel mundial. Todo lo que se va a exponer a partir de ahora proviene del quehacer cotidiano en la Sección de Glaucoma de dicho Hospital donde tuve el privilegio de trabajar durante varios años y ser un eslabón más de la larga cadena de la «Escuela de Glaucoma» del Prof. García Sánchez. Los criterios diagnósticos y terapéuticos, y sobre todo los conceptos, aquí vertidos, son fruto de innumerables charlas, reflexiones y discusiones acumuladas en el tiempo. Aquí van a estar plasmadas las experiencias del Prof. Miralles, el Prof. Zato, el Dr. Fernández Vila y todos aquellos que de una u otra forma nos hemos acercado al problema del glaucoma congénito poniendo nuestro granito de arena, y sobre todo la del Prof. García Sánchez, que ha sido la persona que de forma ininterrumpida ha seguido esta evolución durante todos estos años.
TERMINOLOGÍA Desde que Collins (1) en 1893 y Cross en 1896 (2) determinaron como probable causa del glaucoma congénito la alteración en el desarrollo embrionario de las estructuras del ángulo iridocorneal, han sido muchos los que han llegado a la misma conclusión (3,4), en el sentido que sería una alteración en este desarrollo lo que determinaría una elevación en la presión intraocular y secundariamente el desarrollo de glaucoma. Los llamados glaucomas del desarrollo son un grupo de enfermedades caracterizadas por un defectuoso desarrollo del sistema de drenaje del humor acuoso y aunque el glaucoma puede no manifestarse hasta la edad adulta, la mayoría se presentan en la infancia. El desarrollo incompleto del segmento anterior puede llevar a distintas formas de glaucoma. Con el término de glaucoma congénito se ha designado clásicamente aquellos casos en los que el glaucoma se pone de manifiesto en los primeros años de vida. Si la alteración en el segmento anterior es más leve, provocaría una elevación de la presión más tardía, dando lugar al denominado glaucoma juvenil. Esta terminología ha sido abandonada por muchos autores al considerar que la edad no puede ser criterio para una clasificación (5). Otros autores (4,6) consideran al glaucoma juvenil como un glaucoma congénito tardío que aparece habitualmente después de los 4 años. A esta edad la resistencia parietal del globo ocular es tan similar a la del adulto que ya no se producirían alteraciones morfológicas clínicamente detectables. La clínica y el comportamiento de estos pacientes nos obliga a considerar dos entidades totalmente distintas, que precisan un tratamiento y un seguimiento diferentes en ambos grupos.
I. GLAUCOMA CONGÉNITO I.1. Definición Dentro de los glaucomas del desarrollo, el Glaucoma Congénito Primario es una enfermedad que se manifiesta en los primeros meses de la vida caracterizada por la existencia de una alteración en el desarrollo de la malla trabecular y estructuras angulares, no asociada a otras anomalías oculares o enfermedades sistémicas, y que va a condicionar una elevación patológica de la presión intraocular y secundariamente una lesión glaucomatosa del nervio óptico y alteraciones anatómicas en el globo ocular.
I.2. Epidemiología El glaucoma congénito primario presenta una incidencia de 1/10.000 a 15.000 recién nacidos (0,01 a 0,04%) según los diferentes estudios (7-9); alcanzando incluso 1 de cada 1.250 en la población gitana de Eslovaquia (10). Si se considera el glaucoma infantil (incluyendo glaucomas complicados) la incidencia alcanza el 1/2.000. En un estudio español analizando 1.124.654 nacimientos consecutivos para estudiar las malformaciones oculares congénitas, la prevalencia del glaucoma congenito en el naciemiento es de 2.85/100.000 (11). Aparece de forma bilateral en el 75-80% de los casos, existiendo una predilección por el sexo masculino del 53% al 68% en USA y Europa (7-9,12-14), mientras que en el Japón (8) existe un predominio de mujeres (63%). La mayoría (más del 80%) se manifiestan antes del año de edad.
I.3. Herencia Habitualmente la aparición del glaucoma congénito es esporádica. En el 10% existen antecedentes familiares con un patrón de herencia autosómico recesivo (8,13,14), aunque la penetrancia es muy variable (40-80%) y se cree que se debe a un patrón multifactorial. En algunas familias la aparente transmisión vertical se puede explicar por pseudo-dominancia. La frecuencia de los casos esporádicos se puede ser debida a la falta de penetrancia (40%), la rareza del gen, las nuevas mutaciones y la existencia de fenocopias (13). La asociación del glaucoma congenito con anomalías cromosómicas es muy frecuente, habiéndose reconocido en más de 17 autosomas diferentes (16). Desde el punto de vista del consejo genético podría estimarse el riesgo de presentar glaucoma congénito para los hermanos o descendientes de un niño afectado en el 3%-4%. La forma autosómica recesiva del glaucoma congénito primario (símbolo del gen GLC3) se ha mapeado recientemente en dos loci diferente, GLC3A en 2p21 y GLC3B en 1p36, respectivamente, en familias de origen turco, gitanos eslovacos y de Arabia Saudita (tabla 1) (16). Las mutataciones del gen del citocromo P4501B1(CYP1B1) (17), se han identificado como la causa principal de la enfermedad (18), vinculando el glaucoma congénito primario al locus GLC3A en el cromosoma 2p21. Podría ser que los miembros de la familia del citocromo P450 podría controlar los procesos de crecimiento y diferenciación (19-21). Las mutaciones en el 1p36 aún no se han identificado. Con el extraordinario progreso que se ha realizado en el desarrollo de la genética en esta enfermedad, en un futuro muy próximo se podrá determinar el riesgo individual de una familia, permitiendo un seguimiento efectivo y la instauración de un tratamiento precoz (22). Las goniodisgenesias se heredan de forma dominante (23) y en la misma familia pueden existir glaucomas congénitos puros, tardíos, juveniles y/o congénitos del adulto.
I.4. Clasificación Aunque existen varias clasificaciones de los glaucomas congénitos, la más utilizada es la de Shaffer-Weiss (12) (tabla 2) que los clasifica desde un punto de vista etiológico en:
Aunque frecuentemente se engloban los dos últimos grupos como glaucomas complicados.
Hoskins y Shaffer en 1984 (24) presentan una nueva clasificación basándose en las características anatomopatológicas observadas en 250 casos de glaucoma infantil, según que la estructura más afecta de la cámara anterior sea la malla trabecular, el iris o la córnea. Esta clasificación permite una mayor precisión en la descripción de los casos y permite predecir un pronóstico quirúrgico, además de conseguir una estandarización de la terminología (tabla 3). En esta clasificación se distingue: a) Trabeculodisgenesias aisladas: presentes en aproximadamente el 50% de los glaucomas infantiles y juveniles. No existen alteraciones en la córnea o iris salvo las secundarias a la hipertensión ocular prolongada. b) Iridodisgenesias: además de las alteraciones referidas, pueden verse en el ángulo procesos, vasos anómalos o adherencias iridocorneales. En el iris podemos encontrar:
c) Corneodisgenesias: a parte del aumento de tamaño y opacidad secundarias al aumento de tensión ocular pueden existir defectos en la córnea periférica, medioperiférica o central o bien anomalías en su tamaño. En la mayoría de los casos se asocian defectos iridianos. El glaucoma en estos casos suele tener peor pronóstico.
I.5. Clínica Los síntomas clásicos presentes en el glaucoma congénito son epífora, fotofobia, blefarospasmo; y los signos observables, el aumento de tamaño y la opacidad de la córnea en fases más avanzadas; siendo este último el signo clínico inicial más frecuente y la causa más habitual de consulta, ya que la megalocórnea y la epífora pueden pasar desapercibidas en muchos casos. En el ojo del niño la presión intraocular elevada sobre un globo con menor rigidez escleral va a provocar una distensión generalizada del mismo que se traducirá en alteraciones a nivel de las distintas estructuras oculares. En la córnea se van a producir unas rupturas en la membrana de Descemet (estrías de Haab), que se manifiestan como aumento súbito del edema corneal, con aumento del lagrimeo y epífora. Pueden ser únicas o múltiples y se visualizan como líneas paralelas en la parte posterior de la córnea. Más tarde, el endotelio se extiende sobre ellas desapareciendo el edema. Pueden ser asintomáticas o provocar un astigmatismo irregular que afecte en mayor o menor medida la visión. A nivel del limbo y córnea periférica se produce un crecimiento compensatorio del tejido epiescleral y conjuntival, que adopta la forma de una semiluna blanquecina evidente sobre todo en el limbo superior. Si la hipertensión ocular no se controla, la clínica empeorará y el aumento de tamaño de la córnea y las rupturas en la membrana de Descemet darán lugar a la cicatrización, erosiones y finalmente a ulceraciones de la córnea. La afectación de la zónula en forma de distensión y ruptura puede dar lugar a la subluxación del cristalino. Como consecuencia de todas estas alteraciones anatómicas estos ojos quedan con una debilidad manifiesta de forma que si estos ojos sufren traumatismos contusos, fácilmente se pueden producir hifemas e incluso rupturas del globo, llegando en último término a la ptisis bulbi (25,26). A partir de los 3-4 años, el crecimiento del globo ya no suele progresar pero la esclera sigue conservando su elasticidad (se estima que hasta los 10-11 años) pudiendo dar lugar a una miopía rápidamente progresiva, que debe considerarse como un signo sospechoso de glaucoma (aunque la progresión de la miopía simple es frecuente en el niño), siendo más significativo cuando se trata de una anisometropía (27). La aparición de un nistagmus, es poco frecuente en el glaucoma congénito y su existencia es la expresión de un déficit visual grave. Aunque el niño mayor puede quejarse de dolor, esto es excepcional. Habitualmente, cuando el inicio de la hipertensión ocular secundaria a glaucoma congénito se produce a partir de los tres años de edad no existen síntomas hasta que la pérdida de campo visual se hace evidente.
I.6. Diagnóstico El diagnóstico de presunción se basa en la clínica y en este sentido es primordial la colaboración del pediatra como primer eslabón de la cadena diagnóstica. La inespecificidad de la clínica hace que la sospecha del cuadro sea fundamental para el inicio del examen diagnóstico concreto. La confirmación se basará en la determinación de la presión intraocular (P.I.O.) y en la presencia de signos específicos. Aunque la mayoría de los autores consideran que la P.I.O. en el niño varía en una rango similar a la del adulto, diversos estudios como los de Sampaolesi (28) y Domínguez (29) demuestran que es alrededor de 5 mm Hg inferior a la del adulto. La edad del niño es un factor claramente diferenciador de la estrategia diagnóstica a utilizar. En la mayoría de los casos, los niños mayores de 5 años, y si son especialmente cooperadores a partir de 3 años, pueden ser explorados en consulta sin problemas, de forma que el examen sin anestesia puede darnos el diagnóstico con un alto grado de certeza. Es posible realizar varias exploraciones importantes como: la evaluación de la función visual, biomicroscopia, oftalmoscopia, retinoscopia e incluso la tonometría con un aparato manual (el sistema de tonometría Tono-pen y los pneumotonómetros manuales pueden resultar muy útiles). En el glaucoma congénito primario la P.I.O. suele estar claramente elevada, oscilando entre 30-40 mm Hg, aunque siempre una P.I.O. de 20 ó más mm Hg debe ser considerada como altamente sospechosa. Las exploraciones en la sospecha de glaucoma congénito están condicionadas por el hecho de ser necesario realizarlas bajo anestesia general. En este sentido es importante tener preparado el protocolo quirúrgico para que, si se confirma el diagnóstico, se proceda inmediatamente y en la misma anestesia a la intervención quirúrgica. La secuencia diagnóstica bajo anestesia general es: 1. Medida de los diámetros corneales: la determinación del diámetro corneal es una medida del grado de distensión secundaria a la hipertensión ocular. Los valores normales varían en función del crecimiento normal del globo ocular por lo que sus valores se modifican de acuerdo a la edad del niño: en el recién nacido el diámetro normal horizontal es de 9,5-10,5 mm, a los 5-6 meses es de 11 mm, puede llegar a 11,5-12 mm al año de edad, y como máximo puede medir 12,5 mm a los 3 años. La medida debe considerarse desde el punto en que aparecen las primeras fibras blancas en el limbo hasta el punto opuesto en el limbo del otro lado. Es fundamental realizar una buena medida inicial que nos sirva de referencia para el control posterior. Se considera como medida más fiable el diámetro horizontal ya que, la determinación del diámetro vertical es menos definitoria al existir un ensanchamiento del limbo da lugar a un embriotoxon anterior muy pronunciado en la zona superior, con lo que la medición es más inexacta. Se consideran sospechosas de glaucoma congénito medidas de 12-12,5 en niños menores de 1 año y de 13 mm o más a cualquier edad. A partir de la primera medición, el tamaño de la córnea aporta información no sólo para el diagnóstico, sino también para el seguimiento del control del proceso. El aumento progresivo del diámetro corneal indica control inadecuado del glaucoma, independientemente de la determinación de la PIO, aunque hay que tener en cuenta que cambios inferiores a 0,5 mm no son valorables.
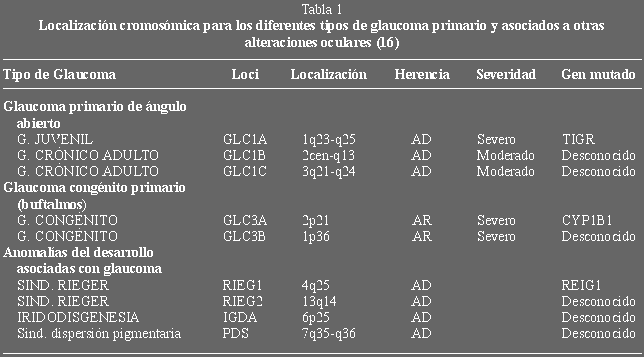
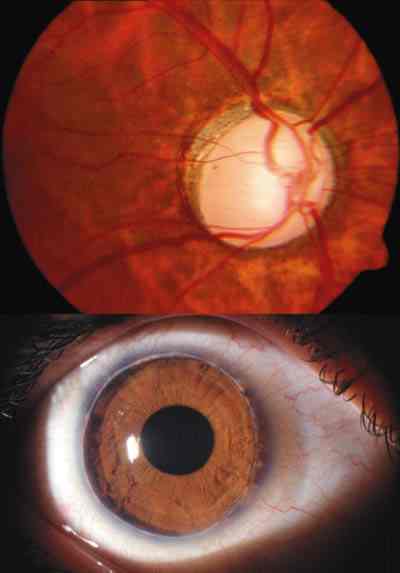
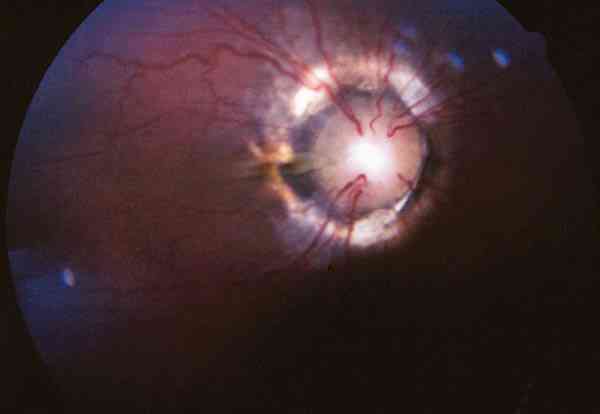
2. Exploración de segmento anterior: debe realizarse con lámpara de hendidura si es posible, y si no, con una linterna y una lupa. Hay que explorar la córnea valorando el grado de transparencia, la existencia de estrías y/o roturas de la capa de Descemet y el grado de edema. En fases más avanzadas la córnea puede tener un aspecto opalescente o puede mostrar zonas opacas en las áreas correspondientes a roturas de la Descemet. Hay que identificar la presencia de alteraciones asociadas en iris o córnea. Esta exploración tiene también valor pronóstico ya que permite valorar el abordaje quirúrgico que podrá realizarse en función de la transparencia de medios (foto 1). En aquellos casos en los que la transparencia de los medios no permita la exploración los nuevos biomicroscopios ultrasónicos puede ser de gran utilidad (30).
3. Gonioscopia: por las características del glaucoma en el recién nacido en ocasiones es difícil de valorar por la pérdida de transparencia corneal. Si la turbidez corneal dificulta la gonioscopia puede mejorarse la transparencia eliminando el epitelio corneal o aplicando glicerina tópica preoperatoriamente. Cuando esto sucede es importante dejar esta exploración para el final ya que impediría una correcta determinación de los otros exámenes diagnósticos.
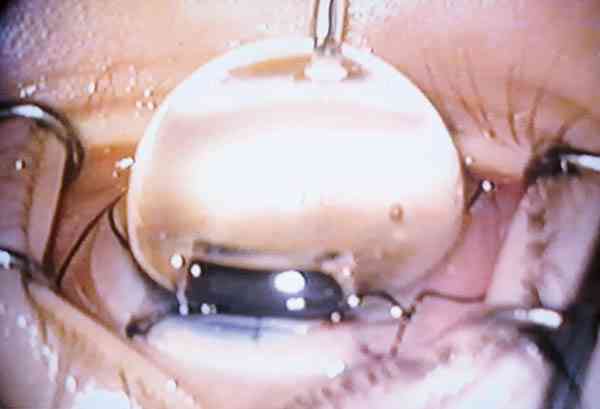
Puede utilizarse cualquier lente de gonioscopia de las usadas habitualmente en la clínica (Goldman, Koeppe, Ritch, etc.). La lente de Worst tiene la ventaja de que proporciona la misma visualización del ángulo que se obtendrá en el momento operatorio por lo que aporta también información de las posibilidades quirúrgicas en caso de goniotomía (foto 2 ).
La apariencia del ángulo en el recién nacido y niño pequeño tiene unas características propias y es distinta a la del adulto. Durante los cuatro primeros años de vida postnatal, continúa el crecimiento de los componentes del ángulo. Así en el niño, el iris en la periferia presenta un estroma anterior delgado, especialmente en los ojos azules, de forma que las arcadas pigmentadas son fácilmente visibles; esto no es una hipoplasia del estroma anterior sino un desarrollo incompleto que posteriormente terminará. Las capas uveal y córneo-escleral de la malla trabecular han aumentado de tamaño, longitudinalmente, mientras las células de la raíz del iris permanecen con una configuración reticular (25). La inserción del iris es normalmente plana, anclándose en el ángulo detrás del espolón escleral, dejando expuesto el cuerpo ciliar. Es raro observar en un ojo normal que el iris se ancle en la malla trabecular o anterior al espolón escleral. En la pared externa del ángulo vemos la línea de Schwalbe, el trabeculado escleral, de color gris un poco más oscuro, el espolón escleral y la banda del cuerpo ciliar de un color más oscuro. En la pared interna vemos la base y raíz del iris. Cuando existe un retraso en la separación del iris de la pared angular puede ser el resultado de una trabeculodisgenesia, siendo este grado de separación lo que distingue una configuración normal u anómala. Es característico ver una especie de triángulos oscuros de base externa que corresponden a la capa pigmentaria del iris, que se transparenta por el escaso desarrollo del estroma iridiano anterior. Separando estos triángulos existen unos cordones más claros que corresponden al tejido conectivo que acompaña los vasos radiales del iris. Dentro de los triángulos pueden verse cordones no vasculares. En la base más externa de los triángulos existe una zona más clara rosada que corresponde al tejido conectivo del círculo arterial mayor del iris y que forma una banda más clara y rosada justo por detrás de la banda ciliar. Pueden existir restos del tejido mesodérmico que le dan al ángulo una apariencia aún más redondeada y que se ven como una fina membrana transparente y de superficie granular, pero sólo podremos ver esta estructura si la buscamos. Cuando los restos mesodérmicos están pigmentados o son opacos ocultan las estructuras que se hallan por debajo del mismo, van desde la raíz del iris hasta la pared externa, a veces hasta la altura del canal de Schlemm, en forma de arborizaciones, y corresponden a los procesos iridianos que a veces pueden verse en el adulto. Hay que diferenciar entre la base y la raíz del iris. La base del iris corresponde al nivel de terminación del último pliegue circular del iris. La raíz del iris se extiende desde la base hasta la banda del cuerpo ciliar. Los procesos mesodérmicos normales no pueden avanzar más que hasta la raíz del iris. El receso normal del ángulo se desarrolla progresivamente en los primeros 6-12 meses de vida. Por la gran fragilidad de las estructuras angulares la información obtenida de los exámenes anatomopatológicos no es totalmente fidedigna. Ademas pocas muestras histopatológicas son representativas, pues la gran parte provienen de ojos alterados con intervenciones quirúrgicas y con glaucoma absoluto. Anderson (6) señaló que la mayoría de los investigadores no han clarificado que características morfológicas del ángulo camerular, en los ojos con glaucoma congénito, son propios de ojos infantiles y cuáles son las que distinguen el glaucoma congénito del ojo infantil normal. En 1940 Castelli describió la histología del ángulo irido corneal de un niño de dos meses, con glaucoma congénito no intervenido. Observó que el canal de Schlemm estaba alterado y parcialmente ausente, el espolón escleral era hipoplásico; la malla uveal era abundante, con una superficie cóncava hacia la cámara anterior. La malla córneo-escleral era compacta y pobremente desarrollada. Había una hipoplasia de la uvea (2). Barkan (3) indicó que la membrana que tapiza normalmente el ángulo, en el glaucoma congénito se hace menos transparente, e impermeable, denominándose membrana de Barkan y ésta impediría el flujo del humor acuoso. Son muchos los autores que han observado esta membrana, sin embargo la naturaleza de ésta es muy discutida y muchos autores cuestionan su auténtica existencia. Anderson (6) 1971, mostró mediante scanning y microscopia de transmisión, que la membrana descrita en estos pacientes, representa las capas más internas de la malla uveal. Muchos autores han observado la configuración fetal del ángulo camerular en el glaucoma congénito, formada por un tejido mesodérmico embrionario idéntico a aquel que constituye el trabeculum primitivo de los ojos de los fetos (23). Sampaolesi (28) indica que la histología del glaucoma congénito presenta el mismo aspecto que el seno camerular en el séptimo mes de desarrollo intrauterino. Por todo lo expuesto la configuración del ángulo camerular del glaucoma congénito es un tema muy controvertido y en resumen podemos considerar patológicos los siguientes hallazgos gonioscópicos (25):
4. Tonometría bajo anestesia general: éste continúa siendo un tema controvertido. Cualquier sustancia farmacológica utilizada en la inducción de la anestesia general va a influir en mayor o menor grado en la presión intraocular. No deben darse barbitúricos o narcóticos previamente porque bajan la P.I.O., la única premedicación aconsejable es la atropina. Puede usarse la ketamina o anestésicos inhalatorios con o sin intubación. La ketamina aumenta la PIO. Los anestésicos inhalatorios bajan la P.I.O. proporcionalmente al grado de profundidad de la anestesia. Es fundamental que en cada exploración se utilice siempre la misma pauta de inducción a la anestesia para que las cifras de PIO obtenidas puedan ser valorables. Podemos considerar como cifras de referencia, aunque luego siempre hay que individualizar cada caso, las siguientes:
En caso de disparidad entre la cifra de presión intraocular y el resto de la exploración hay que tener en cuenta que la determinación de la presión intraocular, sobre todo condicionada por el hecho de ser tomada bajo anestesia general, no es un valor absoluto y hay que considerar factores asociados (31). Así en un estudio realizado por la Academia de Oftalmología de Nueva Orleans, comparando los resultados de la PIO en niños normales y glaucomatosos se encontró (32): Glaucoma (n=169) Normales (n=74) < 21 mmHg 14 < 15 mmHg 26 21-24 mmHg 10 15-21 mmHg 39 > 24 mmHg 145 21-24 mmHg 4 Sin embargo, también hay que tener en cuenta que en la clínica diaria la experiencia nos dice que las cifras de PIO claramente elevadas confirman el diagnóstico de glaucoma congénito.
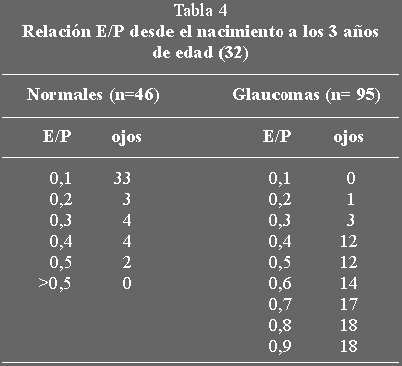
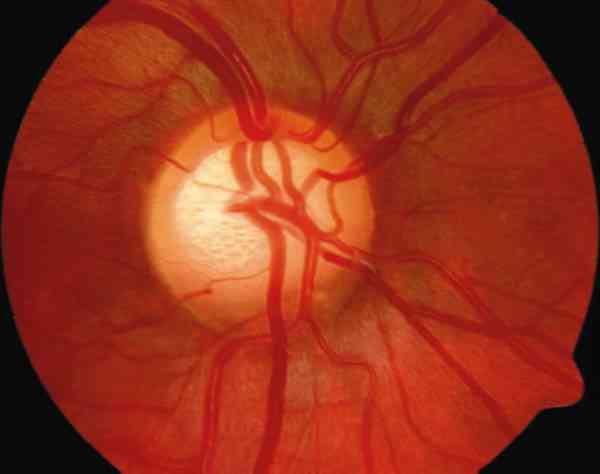
5. Oftalmoscopia: la excavación de la papila es un signo precoz y ocurre antes y con tensiones más bajas que en el adulto, probablemente debido a la mayor elasticidad de los tejidos en el niño y a la falta de desarrollo del tejido conectivo de la lámina cribosa. Robin (33) en su serie de glaucoma congénito observa una excavación media de 0,68±0,24 siendo ésta más acusada verticalmente. Además los cambios en el disco óptico del niño difieren de la de los adultos: el canal escleral se elonga secundariamente a la elevación de la PIO, y el diámetro del canal escleral aumenta más horizontal que verticalmente. Así en estos glaucomas el tamaño de la excavación puede venir determinado por la pérdida de tejido neural, por la elongación del canal escleral o por una combinación de ambos. Quigley en estudios histológicos (34,35), ha observado que mientras el tejido neural, glial y elementos vasculares de la cabeza del nervio óptico tienen la configuración del adulto ya durante la gestación, el tejido conectivo de la lámina cribosa presenta un desarrollo incompleto en el nacimiento.
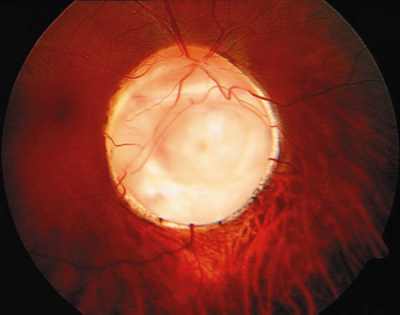
La excavación en el niño suele ser redondeada, muy central y profunda. Se considera patológico una excavación mayor de 0,4, o una asimetría mayor a 0,2 (25) (tabla 4). Cuando la PIO se normaliza, la excavación puede permanecer estable o puede disminuir, de forma muy evidente en el niño menor de 1 año (foto 6).
Por las características de la excavación patológica en el niño su existencia confirma el diagnóstico, pero no guarda relación con la gravedad del proceso ni puede usarse como criterio de control del glaucoma.
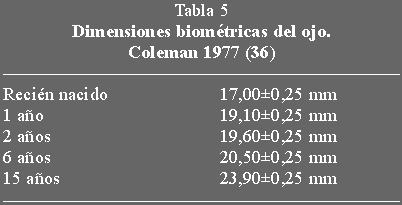
6. Determinación de la longitud axial: Es una exploración fundamental en el glaucoma congénito. Su importancia viene determinada no tanto por el interés en la confirmación del diagnóstico como en el seguimiento del control del proceso glaucomatoso en el tiempo. Tanto es así, que desde nuestro punto de vista es la exploración fundamental en el postoperatorio no pudiendo considerar controlado un glaucoma congénito mientras el eje antero-posterior del ojo siga aumentando independientemente que los valores tensionales puedan parecer normales. Los valores pueden solaparse con los del ojo normal por lo que es necesario establecer una comparación con los valores considerados como normales para cada edad (tabla 5).
7. Retinoscopia: una vez controlada la tensión ocular, la determinación de la refracción bajo cicloplejía es siempre necesaria y especialmente importante en los casos de glaucoma unilateral, para compensar la diferencia de graduación y evitar la aparición de la ambliopía anisometrópica.
8. Exploración sistémica: para detectar posibles anomalías asociadas y como preparación a la anestesia.
I.6.1. Diagnóstico diferencial I. Otros glaucomas
II. Causas de aumento del tamaño corneal y de edema de la córnea
III. Causas de epífora y/o fotofobia
I.7. Tratamiento I.7.1. Tratamiento médico
No existe ninguna posibilidad de tratamiento médico del glaucoma congénito sea cual sea el fármaco empleado. Solamente se puede recurrir a la utilización de fármacos hipotensores en situaciones especiales y muy concretas y siempre de forma transitoria. Estas situaciones son: — Falta de transparencia de medios por edema corneal en el momento de la sospecha de glaucoma:
— Demora en la cirugía:
— Intervalo entre cirugías:
— Fracaso quirúrgico:
I.7.2. Tratamiento quirúrgico El tratamiento del glaucoma congénito, sea cual sea su forma, es única y exclusivamente quirúrgico. Como se ha señalado en el apartado anterior, actualmente no existe ninguna forma de tratamiento médico de esta enfermedad. Las técnicas quirúrgicas que pueden utilizarse para el tratamiento del glaucoma congénito son:
Cada una de ellas tiene su lugar en la estrategia quirúrgica, aunque existen diferencias muy significativas entre los resultados obtenidos con cada una de ellas. Desde nuestro punto de vista, y desde nuestra experiencia la técnica de elección para el tratamiento del glaucoma congénito es la goniotomía. Esta técnica es la única que actúa directamente sobre la causa de la hipertensión ocular en el recién nacido y al mismo tiempo es la que presenta unos mejores resultados a largo plazo en el control de la enfermedad. Sin embargo, el estado del ojo en el momento del diagnóstico de glaucoma congénito va a hacer que en ocasiones el abordaje quirúrgico para la goniotomía no sea posible y sea necesario recurrir a otras técnicas quirúrgicas de segunda elección.
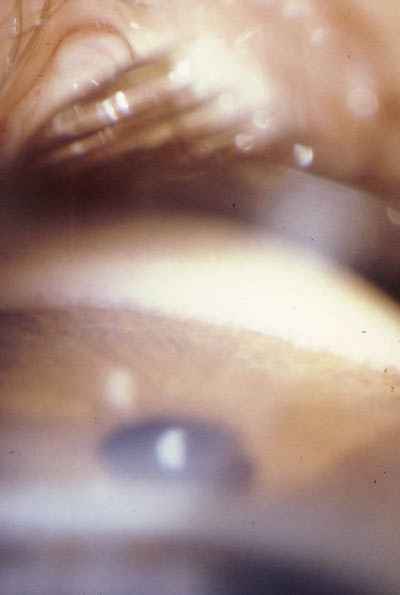
Goniotomía La técnica quirúrgica de la goniotomía está muy sistematizada habiendo presentado pocos cambios desde su primera descripción por Barkan. Los puntos de mayor interés son: Fuente de iluminación: aunque se han utilizado diversas formas de iluminación del campo quirúrgico, la única fuente de luz que permite una correcta visualización y una magnificación suficiente del ángulo iridocorneal es la luz del microscopio coaxial. Las únicas limitaciones que presenta son la necesidad de una suficiente transparencia corneal, la capacidad del microscopio de modificar su ángulo de incidencia sobre la córnea y el hecho de que el cirujano debe trabajar más alejado del campo quirúrgico. Solo en caso de no poder realizar la goniotomía con el microscopio quirúrgico puede recurrirse a la utilización como fuente de luz de un oftalmoscopio binocular indirecto. Gonioscopio: es quizá la parte fundamental en la goniotomía ya que va a permitir la correcta visualización del campo quirúrgico durante toda la cirugía. Existen diversos modelos en el mercado y cualquiera de ellos reúne las condiciones necesarias para la correcta visualización del ángulo, sin embargo nosotros preferimos el gonioscopio fenestrado de Worst ya que permite un campo amplio de visualización de las estructuras angulares y al mismo tiempo el hecho de estar fijado al globo ocular, lejos de ser un inconveniente, supone la ventaja de permitir al cirujano su movilización facilitando las maniobras del goniotomo durante la cirugía. Durante los momentos previos a su colocación sobre el globo ocular es aconsejable mantener el gonioscopio sumergido en una solución que disminuya la tensión superficial con lo que se mejora las características de visualización del instrumento (foto 11). Goniotomo: habitualmente el utilizado es el goniotomo hidrostático de Worst, que permite mediante la irrigación continua el lavado del sangrado que con frecuencia se produce al lacerar los restos mesodermicopretrabeculares vascularizados y al mismo tiempo, aunque en menor medida permite mantener un cierto grado de amplitud de la cámara anterior. Esta circunstancia se ha visto mejorada notablemente con el uso de viscoelásticos durante la cirugía, que posibilitan una buena amplitud de la cámara anterior y una distensión del ángulo facilitando el paso del goniotomo.
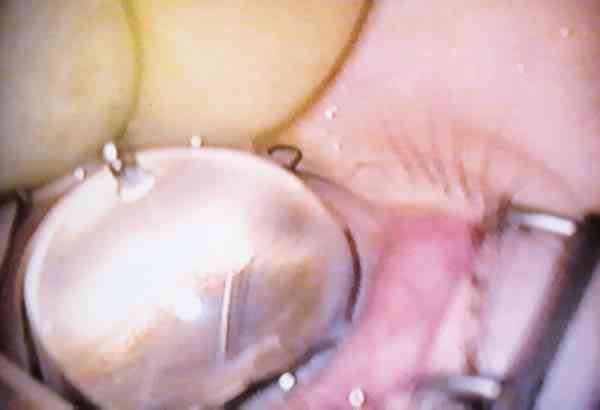
Incisión: la punción con el goniotomo a nivel del limbo esclero-corneal supone a nuestro criterio el punto crítico de la cirugía de goniotomía. Es el único momento en que entramos en cámara anterior con un instrumento punzante «a ciegas» y la mayor atención se centra en la visualización inmediata de la punta del goniotomo en la cámara anterior. Es en este momento quirúrgico cuando pueden suceder las principales complicaciones de la goniotomía derivadas de la lesión directa con el goniotomo de las estructuras de la cámara anterior (endotelio corneal, iris, cristalino) (foto 12).
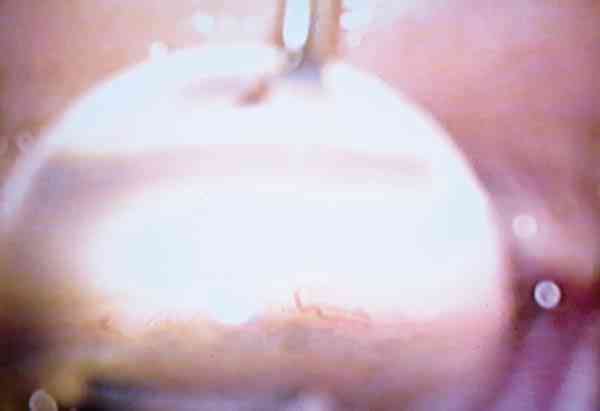
Goniotomía: el otro momento clave durante la cirugía es la colocación correcta de la punta del goniotomo sobre las estructuras angulares de manera que al desplazarse rasge los restos mesodérmicos que tapizan el ángulo y no otras estructuras. Una situación excesivamente anterior, lesionará el endotelio corneal y una muy posterior podrá producir una iridodiálisis. Cuando el goniotomo está correctamente colocado se visualiza una línea de color blanquecino que no es otra cosa que la zona trabecular libre de restos mesodérmicos. Cuando al desplazar el goniotomo se observa movimiento del iris e incluso un cierto grado de plegamiento quiere decir que está situado por debajo de la zona y está desplazando la raíz del iris. El recorrido del goniotomo debe ser tan amplio como lo permita la visualización a través del gonioscopio. Al finalizar esta maniobra se retira el goniotomo, no precisando ninguna sutura corneal, dando por finalizada la cirugía (fotos 13 y 14).
Como preparación a la goniotomía es recomendable la instilación previa de pilocarpina 1% con la finalidad de reducir el tamaño pupilar y ampliar el ángulo iridocorneal. En el niño menor de 3 años con córnea transparente está indicada siempre como primera elección la goniotomía, siendo la técnica preferida por la mayoría de los autores entre 1 y 24 meses de edad, ya que no altera la conjuntiva y actúa únicamente sobre los tejidos del trabéculo superficial. En los niños menores de 1 mes suelen asociarse anomalías del iris que empeoran los resultados, mientras que los mayores de 2 años suelen tener peor respuesta a la goniotomía. En los niños mayores de 3 años, y aquellos menores de 3 años con opacidad corneal que limite la visión del segmento anterior, la trabeculotomía será la técnica de elección y en caso de fracaso de ésta, la trabeculectomía. Es fundamental que el tratamiento quirúrgico se haga precozmente, antes de que la compresión prolongada de las estructuras trabeculares por la hipertensión intraocular impida su reapertura.
Trabeculotomía Al igual que sucede con la goniotomía, es una técnica complicada y require gran experiencia por parte del cirujano. La técnica utilizada es la de Harms (1969) usando el trabeculotomo de dos ramas de Harms para la canalización del canal de Schlemm. El inconveniente de esta técnica es la dificultad de canalizar el canal de Schlemm con el trabeculotomo y en caso de conseguirlo la gran tendencia que tiene a cicatrizar la trabeculotomía realizada. En nuestra corta experiencia con esta técnica los resultados han sido prácticamente nulos por lo que no la utilizamos, sin embargo otros autores reportan resultados satisfactorios en el control del glaucoma congénito (37). En caso de fracaso de ambas técnicas o de ausencia de transparencia corneal, estará indicada la trabeculectomía.
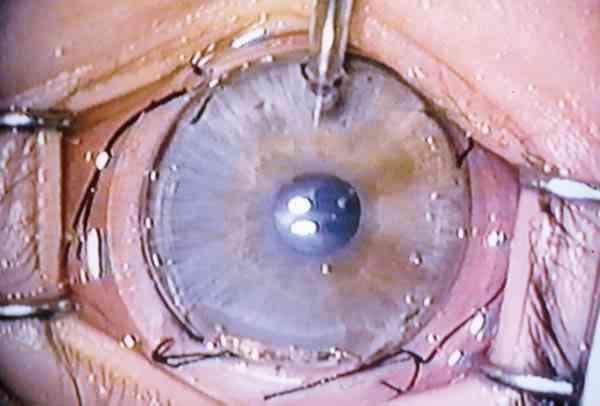
Trabeculectomía La técnica quirúrgica empleada es la misma que la descrita por García Sánchez (38) para el tratamiento quirúrgico del glaucoma primario de ángulo abierto. Las únicas características diferenciales radican en dos hechos (foto 15):
Dado que la principal complicación es el fallo secundario atribuido a un exceso de cicatrización, algunos autores aconsejan el uso de moduladores de la cicatrización. El 5-fluouracilo solo es utilizado de forma preoperatoria de igual manera que la mitomicina C y solo será aplicable en casos seleccionados (39). Algo parecido sucede con la inyección subconjuntival intraoperatoria (antes de abrir la conjuntiva) de un corticoide de acción prolongada como la triamcinolona que puede mejorar el pronóstico. Nosotros no somos partidarios del uso de antimitóticos de primera intención en el tratamiento quirúrgico del glaucoma congénito por los grandes y graves riesgos que puede comportar una dosis inadecuada en una esclera adelgazada y con posibilidad de mayor distensión en el futuro. Solamente en casos de reintervenciones en niños de más de 12 años de edad y en casos muy seleccionados utilizamos mitomicina C. En casos de glaucomas congénitos resistentes al tratamiento y en edades en las que el crecimiento del ojo está estancado preferimos el uso de implantes valvulares tipo Ahmed pediátrico para controlar la hipertensión ocular (foto 16).
ESTRATEGIA TERAPÉUTICA 1. Primera cirugía:
TRABECULECTOMÍA 2. Reintervencion:
TRABECULECTOMÍA + Mitomicina C/5-Fu VÁLVULA DE AHMED PEDIÁTRICA. CONTROL POSTOPERATORIO Tras la cirugía, el primer control se realizará en la primera semana, y después al mes, cada 3 meses durante el primer año y en caso de control del proceso, cada 6 meses hasta los 4 años de edad. A partir de aquí los controles serán anuales. El examen postoperatorio seguirá la misma sistemática que el de diagnóstico de glaucoma congénito. Sin embargo, el valor que cada parámetro de la exploración tiene es diferente. Lógicamente, la cifra de PIO tiene importancia en el concepto de control de la enfermedad pero no es el único ni el más importante. Desde nuestro punto de vista el parámetro más objetivo y menos influenciable por factores ajenos a la enfermedad (como la anestesia general) es la medida del eje antero-posterior del globo ocular. Hasta tal punto esto es así, que con incrementos superiores a 1,5 mm del eje antero-posterior entre exploraciones consecutivas, independientemente de la cifra de PIO consideramos que el glaucoma congénito está descontrolado y requiere nuevamente tratamiento quirúrgico. Cuando el tratamiento inicial ha tenido éxito, el pronóstico funcional en el glaucoma congénito primario es bueno, con buen control tensional, nervio óptico estable y campos visuales habitualmente sin afectación. En los casos en los que la cirugía ha sido tardía o no ha controlado adecuadamente la P.I.O. en los primeros momentos existe un crecimiento marcado del ojo, aunque luego se normalice la PIO. En estos casos, además de la atrofia del nervio óptico y defectos campimétricos secundarios, la distensión del polo posterior puede contribuir a disminuir la visión por distorsión y elongación de la región macular.
I.7.3. Rehabilitación visual Las alteraciones en el tamaño del globo ocular secundarias a la hipertensión ocular en el glaucoma congénito, sobre todo en el aumento del eje antero-posterior del ojo van a condicionar una serie de defectos de refracción más o menos importantes en función de la gravedad del caso. Esto unido al hecho de que habitualmente estos glaucomas son asimétricos y en ocasiones unilaterales condiciona la aparición de anisometropías. La ignorancia de este fenómeno y de su repercusión sobre la función visual, ha dado lugar en ocasiones a déficits importantes en la función visual incluso ceguera legal en casos en los que el control tensional después de la cirugía había sido un éxito. Actualmente no se puede abordar el tratamiento de un glaucoma congénito si no se dispone también de un servicio de rehabilitación visual que coordine el tratamiento de la ambliopía secundaria a la anisometropía resultante en todos los casos.
I.7.4. Resultados En nuestra experiencia tenemos estudiada una muestra de un total de 573 casos diagnosticados de glaucoma congénito y tratados todos ellos quirúrgicamente en un período de tiempo comprendido entre 1969 y 1999 en la Sección de Glaucoma del Servicio de Oftalmología del Hospital Clínico San Carlos de Madrid. En esta muestra el 79% de los casos fueron glaucomas congénitos bilaterales, sin significación estadística en cuanto a la afectación por sexos. Desde el punto de vista gonioscópico se encontró:
La edad media al momento del diagnóstico fue de 2,9±4,58 meses, mientras que la edad media al momento del diagnóstico en nuestro centro fue de 6,8±10,40 meses. Una vez confirmado el diagnóstico la primera cirugía se realizó con una edad media de 8,77±21,95 meses. En algunos casos los pacientes venían previamente diagnosticados de glaucoma congénito y con tratamiento médico previo instaurado en el centro de origen (tabla 6).
En la exploración realizada para confirmar el diagnóstico de glaucoma congénito los parámetros clínicos fueron:
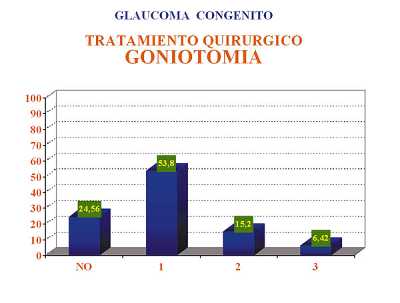
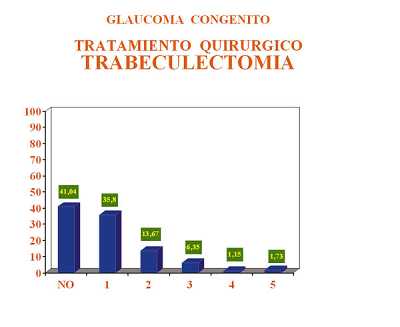
Por las características de los glaucomas en el momento del diagnóstico se pudo realizar goniotomía como técnica de elección en el 63,33% de los ojos y en el 36,66% se tuvo que realizar trabeculectomía por imposibilidad de realizar goniotomía. Si analizamos el número de intervenciones quirúrgicas que fue necesario realizar en los casos tratados con goniotomía y en los tratados con trabeculectomía se observa que fue mayor el número de cirugías en los casos con trabeculectomía primaria. Esto concuerda con el hecho de que los ojos en los que no se puede realizar de primera intención una goniotomía es porque presentan un peor estado de transparencia ocular y consecuentemente se trata de glaucomas más avanzados y la mayoría de las veces de aparición más precoz lo que empeora el pronóstico final (figuras 1 y 2, tablas 7 y 8).
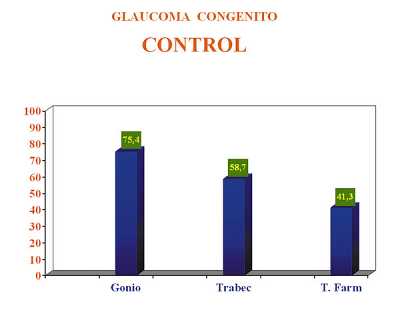
El control conseguido con goniotomía fue significativamente superior al obtenido con la trabeculectomía. En los ojos operados con goniotomía se obtuvo el control del proceso en un 75,40% de los ojos intervenidos. Con la trabeculectomía solo se obtuvo el control en el 58,70% de los casos, siendo necesario en un 41,30% asociar tratamiento médico hipotensor por fracaso de la cirugía. Estos resultados concuerdan con lo expresado anteriormente en cuanto a la gravedad de los casos en función de la técnica quirúrgica empleada (figura 3).
La probabilidad de control de la enfermedad en los ojos tratados aparece reseñada en la figura 4.
La evolución de la PIO en el primer año del postoperatorio con ambas técnicas fue similar, siendo menor la PIO media obtenida con goniotomía (figura 5).
Los datos referentes a evolución del diámetro corneal y de la longitud axial después de la cirugía se reflejan en las figuras 6 y 7 respectivamente.
Las complicaciones quirúgicas con cada una de las técnicas quirúrgicas aparecen reseñadas en la tabla 9.
I.8. Rehabilitación visual Tienen especial importancia como se comentó en el apartado de tratamiento del glaucoma congénito la rehabilitación visual de la ambliopía resultante de la anisometropía secundaria al aumento de la longitud axil del ojo. Los controles realizados a los pacientes se han llevado a cabo con la misma periodicidad que las otras exploraciones, indicando la corrección óptica necesaria lo más precozmente posible después de la normalización de la cifra de presión introcular. La pauta seguida en nuestros pacientes fue la siguiente:
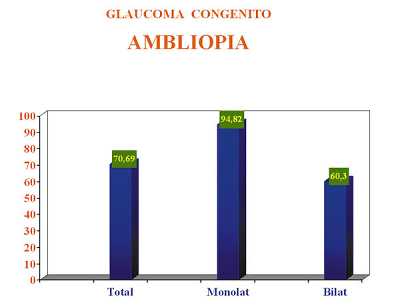
Con esta pauta de tratamiento de la ambliopía las revisiones deben realizarse cada 15 días y se irá modificando la pauta en función de la evolución de cada caso en particular. En nuestros casos, la aparición de ambliopía en mayor o menor grado fue en un 70,69% de todos los casos diagnosticados y tratados. Cuando el glaucoma congénito fue monolateral la ambliopía se produjo en un 94,82% de los ojos, mientras que cuando la afectación fue bilateral la ambliopía apareció en el 60,30% de los casos (figura 8).
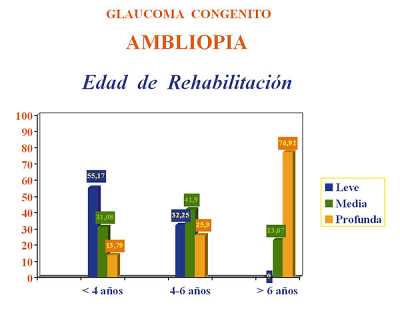
Si analizamos el grado de ambliopía resultante en función de la edad en la que se inicia la rehabilitación después del control del glaucoma congénito se observa que hay diferencias muy significativas entre lo conseguido antes y después de los 4 años de edad. Cuando la rehabilitación visual se inicia antes de los 4 años de edad los resultados de ambliopía leve son moderadamente satisfactorios (55,17%), mientras que a partir de esta edad existe un pequeño porcentaje de ojos con ambliopía leve y sin embargo se incrementa notablemente el número de ambliopía profundas, llegando hasta un 76,93% en mayores de 6 años (figura 9, tabla 10).
Como resumen, se puede concluir que el glaucoma congénito es una enfermedad infantil altamente invalidante, que suele provocar un déficit importante de la función visual a pesar del tratamiento y que la única posibilidad de impedir ó al menos limitar esta evolución hacia la ceguera se basa en los mismos tres pilares que el resto de enfermedades:
AGRADECIMIENTOS Al Dr. P. Gili Manzanaro, de la Fundación Hospital Alcorcón, por su colaboración en la iconografía. BIBLIOGRAFÍA
|