|
INTRODUCCIÓN Morton F Goldberg en el LIV Memorial Edward Jackson (1997) propuso definitivamente el cambio de nombre de la persistencia del vítreo primario hiperplásico, por persistencia de la vasculatura fetal (PVF) al considerar que se integran en su definición y clasificación muchas más entidades de esta compleja patología. Pasarán muchos años hasta que nos acostumbremos a esta nueva denominación, pero tendremos que hacerlo porque la antigua (PVPH) es incompleta. Dentro del diagnóstico diferencial del retinoblastoma y otras causas de leucocoria, uno de los cuadros que se deben tener en cuenta es la persistencia de la vasculatura fetal (PVF), antes llamada persistencia de vítreo primario hiperplásico (PVPH). Descrito por primera vez por Reese (1), consiste en una malformación congénita de la porción anterior del vítreo primario que se presenta generalmente como una placa de tejido conectivo fibrovascular retrolental. Esta estructura, que puede contener tejido adiposo, cartílago y músculo liso (2), además de vasos y fibroblastos, puede adherirse a la cápsula posterior del cristalino conduciendo a la opacidad progresiva del mismo. Por otro lado, esta placa fibrosa, puede extenderse lateralmente hasta unirse con los procesos ciliares que sufrirán una elongación centrípeta. Un cristalino «intumescente» junto con unos procesos ciliares proyectados anteriormente, producen el abombamiento del iris que conduce a la presencia de una cámara poco profunda y al desarrollo de glaucoma que característicamente se presenta asociado al cuadro de PVF.
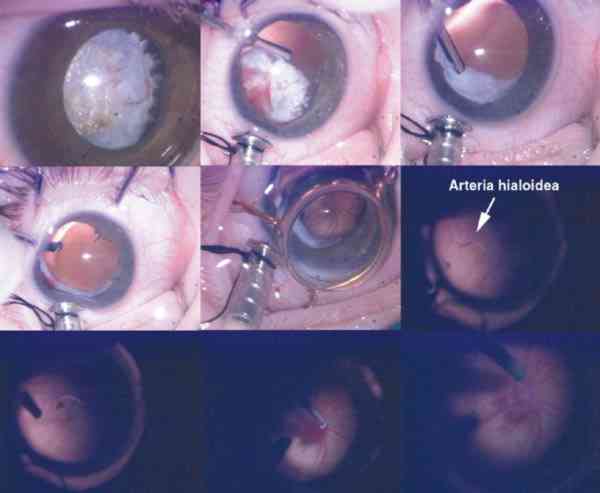
El 90% de los casos son unilaterales aunque con mucha frecuencia se pueden encontrar en el ojo adelfo alteraciones menores en la regresión normal del vítreo (3) como son la mancha de Mittendorf, opacidad nasal inferior en la cápsula posterior del cristalino, recuerdo de la unión durante el período fetal de la arteria hialoidea y la túnica vasculosa lentis, o una papila de Bergmeister, remanente de la porción posterior de la arteria hialoidea.
Los casos bilaterales suelen verse en pacientes con otras anormalidades sistémicas (2) (p.e. trisomía 13, labio leporino, paladar hendido, polidactilia y microcefalia) y asocian generalmente afectación más severa del polo posterior (grados variables de displasia retiniana).
Es posible encontrar PVF en el 3% de los recién nacidos a término, y en algún grado, en el 95% de los prematuros (4). En niños menores de 36 semanas de gestación, en más del 90% de los casos podemos hallar signos que implican una regresión incompleta del sistema vascular hialoideo (5).
CLASIFICACIÓN La PVF se clasifica en una forma anterior, posterior o mixta según donde se encuentre localizada la afectación ocular. Esta distribución tiene una implicación pronóstica ya que son los pacientes con la forma anterior pura los que, tras el oportuno tratamiento quirúrgico, están en condiciones de alcanzar cierto grado de función visual. Los hallazgos exploratorios que caracterizan la forma anterior de la PVF son:
En la forma posterior es posible encontrar:
Lo más común es encontrar formas mixtas de la enfermedad, existiendo en un mismo ojo características tanto de la forma anterior como de la posterior.
DIAGNÓSTICO CLÍNICO Y DIAGNÓSTICO DIFERENCIAL Shields y col (6) publican una serie de más de 100 pacientes con el diagnóstico de sospecha de retinoblastoma. Tras estudiar los casos, se confirma este diagnóstico en el 58% de los pacientes. En el resto, el diagnóstico final fue de PVF en el 16%, enfermedad de Coats en otro 16% y toxocariasis ocular en otro 16%. Howard y Ellsworth (7) estudiaron 500 pacientes diagnosticados de retinoblastoma. En el 53% de los pacientes de la serie hubo que cambiar el diagnóstico. Encontraron un elevado porcentaje de casos de PVF que fue catalogada en un principio como retinoblastoma. Otros diagnóticos fueron fibroplasia retrolental, catarata posterior y uveitis. Las entidades que pueden confundirse con cierta frecuencia con la PVF son:
Enfermedades que raramente pueden ser confundidas con PVF:
Ante la sospecha diagnóstica de PVF se pueden realizar una serie de pruebas complementarias que nos ayuden a definir y delimitar mejor la extensión de la enfermedad así como seleccionar aquellos casos que puedan beneficiarse de un eventual tratamiento quirúrgico. El TAC pretende descartar la existencia de calcio intraocular, hallazgo que de existir, habla muy a favor de la posibilidad de retinoblastoma. No obstante Morris (8) describió calcio en un caso de PVF, aunque esta es una situación muy rara. Otros autores (9) han demostrado la superioridad del TAC frente a la RMN para la detección de calcificaciones. No obstante la RM fue superior al TAC para diferenciar retinoblastoma sin calcio de otras entidades tales como enfermedad de Coats, infección por Toxocara, desprendimiento de retina y PVF. Ante un paciente con opacidad de medios y sospecha de PVF la ecografía puede ayudar a determinar la presencia de enfermedad cuando no es posible hacerlo mediante oftalmoscopía. La ECO-B puede mostrar el tallo vítreo desde polo posterior hasta cristalino y es capaz de poner de manifiesto un posible desprendimiento de retina así como confirmar o descartar la posible microftalmía asociada (10,11). El conocimiento de la longitud axial del globo nos será de utilidad para el cálculo de la LIO a implantar en el caso que decidamos corregir de esta forma la afaquia postquirúrgica. Se ha utilizado también ECO DOPPLER para mostrar la naturaleza vascular de la PVF (12). Por último, la realización de pruebas electrofisiológicas como son los potenciales evocados visuales pondrán de manifiesto la existencia o no de algún resto de función visual, dato que nos puede ayudar en la selección de los casos que se pueden beneficiar de la cirugía (13).
TRATAMIENTO Hoy en día existen controversias sobre que pacientes son candidatos a la cirugía, ya que, a pesar de los avances en el conocimiento de la enfermedad y la notable mejora que ha sufrido el instrumental quirúrgico, los resultados visuales siguen siendo bastante insatisfactorios.
Se podría plantear el tratamiento quirúrgico en pacientes con PVF en los que se desee obtener alguno de estos objetivos: 1. Salvar el ojo de las complicaciones de la PVF no tratada, principalmente el glaucoma (generalmente por cierre angular asociado a un cristalino intumescente tras producirse la rotura de la cápsula posterior por la invasión del tejido fibrovascular) (14) y la ptisis.
Varios trabajos han descrito pacientes con PVF que han llegado a la edad adulta sin necesidad de someterse a la cirugía (15-17). Se ha descrito en algunos pacientes (Goldberg y Peyman) (18) la reabsorción espontánea del cristalino cataratoso, proceso secundario a la reabsorción de la membrana retrolental. Este hecho va a facilitar que el ángulo iridocorneal se mantenga abierto. Bawa y Trese (19) recomiendan un manejo conservador en casos seleccionados, en general pacientes sin potencial visual en el ojo afectado. No obstante, estos autores defienden la cirugía precoz para acortar todo lo posible el período de ambliopía deprivacional. En este sentido, Karr y Scott (20) postulan que el indicador más importante de la función visual futura del paciente, es la edad de presentación del cuadro, ya que estos autores encontraron mejores resultados visuales, tras la cirugía, en los casos de menor edad. 2. Intentar conseguir una pupila "negra", con una apariencia lo más normal posible. Si esto no es posible obtenerlo mediante cirugía, podemos plantearnos, como una alternativa válida, el uso de lentes de contacto cosméticas.
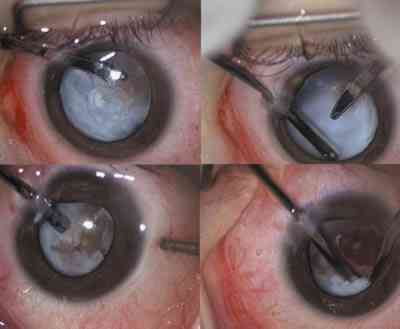
Federman y col (21) propugnan la cirugía precoz para conseguir un mejor resultado cosmético final y obtener un crecimiento del globo ocular más «simétrico». La cirugía de estrabismo también se suele realizar con fines cosméticos ya que el potencial fusional de estos pacientes es muy pobre. No obstante incluso aún de no obtener buena visión tras la cirugía de la catarata monocular, la corrección óptica de la afaquia puede mejorar significativamente la alineación de los ojos (22). 3. En ojos con escasa afectación estructural, es posible la rehabilitación visual después de eliminar el cristalino y las membranas hiperplásicas del vítreo. Como se comentó anteriormente, una vez eliminada la afectación anterior, el pronóstico visual va a depender fundamentalmente del estado del polo posterior. Tan importante es la cirugía precoz, como después de ésta, instaurar una terapia intensiva de oclusiones y corrección óptica para combatir la profunda ambliopía que padecen estos pacientes. Desde la primera descripción de la enfermedad por Reese (1) en 1955 se han intentado muchas aproximaciones quirúrgicas para tratar la PVF. Hoy en día, y gracias al desarrollo del instrumental y equipos quirúrgicos, se hace posible el manejo de la persistencia de vítreo. La lensectomía prevendrá el glaucoma secundario, al suprimir el desplazamiento anterior del diafragma iris-cristalino que puede causar un cierre angular. Eliminando mediante vitrectomía la membrana proliferativa retrolental reducimos la tracción que ésta produce sobre los procesos ciliares, disminuyendo así la posibilidad de que el ojo evolucione hacia la ptisis bulbi. La vitrectomía se realiza por dos o tres vías a nivel del limbo esclerocorneal (19). Las esclerotomías realizadas más posteriores, a nivel de la pars plana-plicata pueden resultar en daño retiniano ya que la retina periférica puede encontrarse traccionada y unida a la membrana retrolental (23). En un trabajo publicado por Federman (21) sobre 15 casos de PVF tratados mediante vitrectomía, realizando las esclerotomías entre 2 y 3,5 mm posterior al limbo esclerocorneal, encontró prolapso retiniano a través de la esclerotomía en un caso, formación de agujeros retinianos yatrógenos al pasar el terminal de vitrectomía por las esclerotomías en otro caso, y formación de una diálisis de más de 200° en un tercer caso. No obstante, en la literatura encontramos otros trabajos que defienden la pars plana-plicata como vía de entrada para la realización de la vitrectomía. Frezzotti (24) utilizó la pars plana, sin complicaciones, en dos casos de PVF. Este autor piensa que, incluso en ojos microftálmicos, realizando las esclerotomías 3 mm posterior al limbo es improbable que se produzca daño en la retina. Una vez eliminado el cristalino, se puede usar diatermia para cerrar los vasos de la membrana retrolental y así disminuir la posibilidad de sangrado durante su disección. En el caso que exista un tallo vítreo central que una la papila con la membrana retrocristaliniana, se puede nuevamente aplicar diatermia a los vasos hialoideos para evitar la hemorragia. En los casos con afectación posterior debemos completar la vitrectomía y realizar pelado de membranas epiretinianas en el caso de exisitir desprendimientos de retina o pliegues retinianos traccionales.
NUESTRA EXPERIENCIA Hemos revisado 25 pacientes (28 ojos) con PVF remitidos, entre los años 1990 y la primera mitad de 2000, a la sección de oftalmología infantil del Hospital La Paz. De los 25 pacientes, 13 fueron niños y 12 niñas. El OD resultó afectado en 11 casos, al igual que el OI. La afectación fue bilateral en 3 pacientes (12%). Según series más amplias publicadas en la literatura (25,26), el porcentaje de bilateralidad oscila entre un 2,5%-11%). Veinte casos tenían menos de un mes de vida en el momento del diagnóstico, en 7 la edad de presentación estuvo comprendida entre 1 y 3 meses, y en un caso el cuadro debutó a los 2,5 años aquejando el paciente pérdida de AV como síntoma principal. El tiempo de seguimiento de los pacientes osciló en un rango de entre 9 meses y 8 años. En 20 pacientes la primera manifestación de la enfermedad o motivo responsable de la consulta y/o derivación del paciente a nuestro servicio fue la leucocoria. El estrabismo fue la segunda causa de consulta (5 pacientes). Como antes se ha señalado, un caso se presentó aquejando disminución de AV.
Encontramos una forma anterior pura de PVF en 6 casos (1 caso de afectación bilateral). En los pacientes restantes se objetivó una forma mixta de la enfermedad (hallazgos de PVF en el polo anterior y posterior del ojo). No encontramos formas posteriores puras. Esta distribución coincide con series publicadas por otros autores (19). En todas ellas se demuestra la mayor frecuencia de presentación de las formas mixtas de la enfermedad. Recordemos que es importante categorizar al paciente según la localización del proceso ya que, en principio, son los casos con PVF anterior los que poseen mayores posibilidades de obtener un mejor resultado visual. Entre la patología posterior encontrada en nuestros pacientes, destaca 1 caso bilateral con hallazgos de displasia retiniana; en dos casos se apreció la existencia de un pliegue retiniano. En uno de ellos, este pliegue ponía en contacto la retina nasal con la masa retrolental. No obstante, el hallazgo más común en las formas mixtas fue la presencia de un tallo vítreo central que contenía restos de la arteria hialoidea. Generalmente este tallo unía la masa retrocristaliniana con la papila, a excepción de un caso en el que el tallo vítreo se insertaba en retina directamente, muy cerca de la mácula del paciente. Tomando como referencia una longitud axial normal al nacimiento de 16,78 mm de media (DS 0,51 mm) (27), encontramos en nuestra serie microftalmos en 10 ojos (dos casos bilaterales). En dos pacientes, aunque el ojo afectado tenía un diámetro anteroposterior en el límite normal (16,3 y 16,8 mm), lo consideramos como un microftalmos relativo al tener los ojos adelfos una longitud axial de 19 y 21 mm respectivamente. Nuestro porcentaje de microftalmía (35,7%) difiere del encontrado por otros autores. Así, Haddad y col (25) encontraron un 66% de ojos microftálmicos en su serie (62 casos). Con excepción de un caso, todos los pacientes con PVF bilateral tenían ojos extremadamente pequeños. En nuestra serie, de los 3 pacientes con afectación bilateral, en dos de ellos encontramos microftalmía bilateral.
Apreciamos 3 casos de microcórnea (diámetro corneal al nacimiento menor de 10 mm), dos asentaban en ojos microftálmicos (caso de afectación bilateral). En otro paciente con microcórnea, el globo ocular tenía un eje anteroposterior de 20 mm. Aunque ciertamente la muestra es pequeña, parece confirmarse la afirmación de Pollard de que la presencia de microcórnea en un ojo con PVF normalmente se acompaña de microftalmía. En nuestra serie no hemos encontrado ningún tumor intraocular acompañando a la PVF. En diferentes publicaciones se ha descrito diferentes asociaciones. Así se ha encontrado un adenoma del epitelio pigmentario del cuerpo ciliar en un ojo enucleado con PVF (28). Shields (29) publicó 1 caso de meduloepitelioma maligno teratoide del cuerpo ciliar que se presentó con características típicas de PVF (leucocoria, cámara anterior estrecha, catarata y membrana fibrovascular retrolental). En los casos de afectación bilateral (3 pacientes) no hemos hallado alteraciones sistémicas asociadas a la PVF. En sólo un paciente con afectación de ambos ojos se descubrieron en la exploración características compatibles con displasia retiniana. Este dato podría apoyar la observación defendida por varios autores de que la PVF bilateral se presenta frecuentemente con afectación más severa del polo posterior. Patologías que se han asociado con la PVF ya se presente ésta de forma uni o bilateral son la trisomía 13, el síndrome de Aicardi, la incontinentia pigmenti, displasia retiniana, síndrome de Morning Glory, retinopatía de la prematuridad, síndrome de Walker Warberg, enfermedad de Norrie (19,30-36). Bawa Das y Trese (19,37) apuntan una posible relación entre el abuso de cocaína durante el embarazo y el desarrollo de PVF. Aunque se han descrito casos de PVF en la misma familia (38), no hemos encontrado ningún caso de agregación familiar en nuestra serie. En todos los pacientes se realizó ecografía ocular como método auxiliar de diagnóstico, para medir el eje anteroposterior del globo ocular, y como medio de valoración del estado del polo posterior en aquellos casos donde la opacidad de medios impedía el estudio oftalmoscópico. Se realizó TAC en algún caso para descartar la presencia de calcio intraocular. Planteamos la cirugía en todos los casos realizando, en los pacientes con afectación anterior pura, lensectomía más vitrectomía vía limbal con extirpación de la membrana fibrovascular, liberándola de sus adherencias a los procesos ciliares, con o sin diatermia de los vasos hialoideos. En los casos de PVF con afectación posterior se eliminaba mediante vitrectomía el tallo vítreo central realizando en ocasiones diatermia de la arteria hialoidea. En sólo un paciente con afectación bilateral (displasia retiniana) se desestimó la cirugía del segundo ojo, dada la mala evolución postquirúrgica del primero, y la nula esperanza de mínima recuperación visual en el ojo no intervenido. En cuatro casos no fue posible realizar seguimiento del paciente ya que, tras la cirugía, los padres no volvieron a revisión. En otro paciente con una forma mixta de la enfermedad, desprendimiento de retina asociado y afectación macular, los padres desestimaron cualquier intento terapéutico dado el mal pronóstico del caso. Las reintervenciones se debieron fundamentalmente a la aparición en 4 casos de membranas inflamatorias postquirúrgicas que ocluían el eje visual y que debieron ser eliminadas mediante cirugía; a dos pacientes se les sometió a una trabeculectomía para controlar aumentos de la tensión ocular secundarias a sangrados intraoculares repetidos y al desarrollo de sinequias iridocapsulares. Finalmente tuvimos que implantar en estos dos casos, válvula de Ahmed para controlar la tensión ocular. Otros 7 pacientes desarrollaron, durante el seguimiento, endotropía que requirió corrección quirúrgica. Se optó por el implante de LIO secundaria en 4 pacientes que no toleraron la lente de contacto, método de elección, en principio, para corregir la afaquia postquirúrgica. Sólo en dos casos se colocó de entrada lente intraocular. Uno de ellos fue un paciente con el diagnóstico de catarata congénita y al que se encontró en el quirófano, tras extraer el cristalino, una pequeña placa de tejido fibroso retrolental que fue eliminada en el mismo acto quirúrgico. Tres pacientes desarrollaron un desprendimiento de retina tras la cirugía de PVF. En dos casos se recomendó la realización de nueva vitrectomía con tamponamiento de silicona. Los padres rehusaron la reintervención. El tercer paciente se sometió a cirugía escleral clásica con buen resultado anatómico y funcional. Otro caso desarrolló sangrados repetidos con hemorragia vítrea organizada que requirió vitrectomía. Los padres también rehusaron la intervención. En un caso hubo que enuclear el globo ocular por evolucionar, tras la cirugía, hacia la ptisis bulbi. En este paciente se colocó una prótesis de hidroxiapatita. En ningún caso hemos tenido problemas con la localización de las esclerotomías para la vitrectomía, no apreciando daño yatrógeno en la retina periférica por este motivo. Se instauró tratamiento corrector de la ambliopía mediante oclusión del ojo director. El modo preferido de corrección óptica fue la lente de contacto, dejando el implante secundario para aquellos casos donde no se pudo colocar la lentilla. Se utilizaron gafas en algunos casos. Diferentes autores han recomendado un tratamiento agresivo de la ambliopía para intentar recuperar cierto grado de visión en ojos con PVF. Pollard sólo recomienda tratar los casos de afectación anterior pura ya que él encuentra muy pobres resultados visuales en los casos con afectación posterior. Globalmente hemos encontrado tras la cirugía, visiones iguales o mayores de 0,1 en el 20% de los ojos. En el 10% de los casos obtuvimos visión de percepción de luz. En otro 10% de los ojos el resultado final fue de no percepción luminosa. En el resto de casos no fue posible completar el estudio por falta de seguimiento. En dos pacientes se ha constatado, con el paso del tiempo, una miopización considerable y progresiva del ojo afectado. Uno de los casos sufrió implante secundario de lente intraocular de +22 D. En la actualidad su vision es de 0.1 corregido con una lente de contacto de –11 D. Estos resultados se asemejan a los publicados en la literatura por otros autores. Pollard (26) obtiene un 18% de visiones igual o mejores de 20/200. Trese (19) presenta una serie en la que el 17% de sus pacientes mantienen algún grado de AV con la carta de Snellen a pesar de la afectación del polo posterior. Dado que la gran mayoría de los pacientes con PVF nunca obtendrán una visión útil existe hoy en día gran controversia sobre que pacientes deben ser sometidos a la cirugía. Parece claro que los casos con sólo afectación anterior se beneficiarán más de una intervención precoz que elimine prontamente la opacidad de medios que conduce a la ambliopía deprivacional. En las formas mixtas o con gran componente posterior, la recuperación visual va a depender enormemente del grado de alteración retiniana. Algunos autores propugnan la realización prequirúrgica de pruebas funcionales como los potenciales evocados visuales en un intento de discernir el potencial visual del paciente. En los casos muy afectados se puede plantear una actuación conservadora. Cuestiones cosméticas o para prevenir el desarrollo de un posible glaucoma o de complicaciones hemorrágicas que desemboquen en ptisis, pueden ayudarnos a tomar la decisión quirúrgica.
BIBLIOGRAFÍA
|