|
ENFERMEDAD DE COATS
Introducción George Coats (1) fue el primero en describir una enfermedad retiniana idiopática que posteriormente llevaría su nombre y se caracteriza por exudación masiva intra y subretiniana con anomalías vasculares periféricas, que se presenta generalmente de forma unilateral en varones jóvenes, sin evidencia de enfermedad sistémica. Coats separó esta nueva entidad en tres grupos: 1) ojos con exudación subretiniana masiva sin anomalías vasculares demostrables; 2) ojos con exudación subretiniana masiva y múltiple anomalías vasculares retinianas con hemorragias retinianas; 3) ojos con malformaciones arteriovenosas francas y exudación subretiniana. Este tercer grupo sería separado después por Von Hippel al demostrar que se trataba de una enfermedad diferente caracterizada por una angiomatosis de la retina. Posteriormente Leber describió una enfermedad con alteraciones vasculares similares a la enfermedad de Coats pero sin la existencia de una exudación subretiniana masiva pasándose a llamar más tarde Enfermedad de Múltiples Aneurismas miliares de Leber. Reese en los años 50 aclaró la situación demostrando que la enfermedad de Leber era una manifestación inicial y parte del síndrome de Coats al observar que un paciente con enfermedad de Leber en su evolución desarrolló una enfermedad de Coats (2).
Etiopatogenia La etiología de esta enfermedad está poco clara. En el pasado muchos investigadores estudiaron la posibilidad de que fuera de origen infeccioso, inflamatorio, metabólico y de otros orígenes sin haberse podido nunca demostrar una relación consistente con ninguna de estas causas (2). En ocasiones ha sido descrito como un síndrome en asociación con varias enfermedades oculares hereditarias como Retinitis Pigmentosa , síndrome de Hallermann Streiff, Síndrome de Senior Loken, Síndrome de Cornelia de Lange, Síndrom de Parry Romberg y el síndrome del Nevus epidérmico (3-8). Últimamente Black et al (9) describen una mujer con enf. de Coats, que dio a luz a un niño con enfermedad de Norrie, portando ambos una mutación en el gen NDP en el cromosoma Xp11.2. Posteriormente analizaron las retinas de 9 ojos enucleados de varones con enf. de Coats, hallando en uno una mutación somática en el gen NDP que no estaba presente en tejido no retiniano, por lo que sugieren, que la enfermedad de Coats es secundaria a una mutación somática del gen NDP que resulta en una deficiencia de Norrin (proteína que fabrica este gen) dentro del desarrollo retiniano, que además, como confirman otras observaciones esta proteína sería crítica también para una vasculogénesis normal (10).
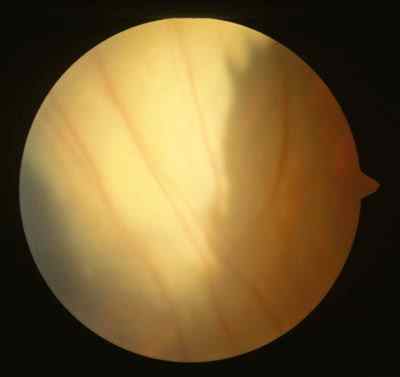
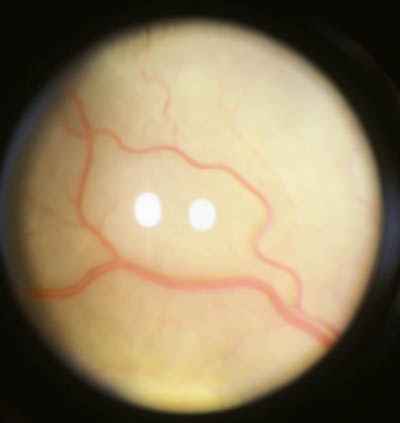
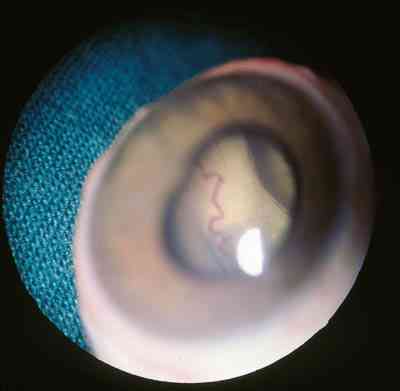
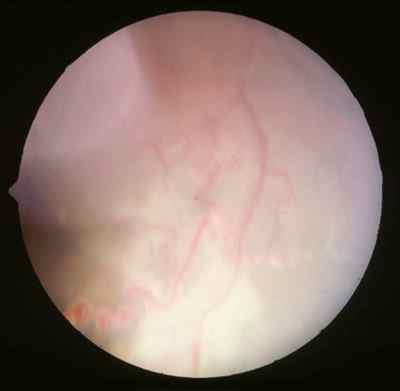
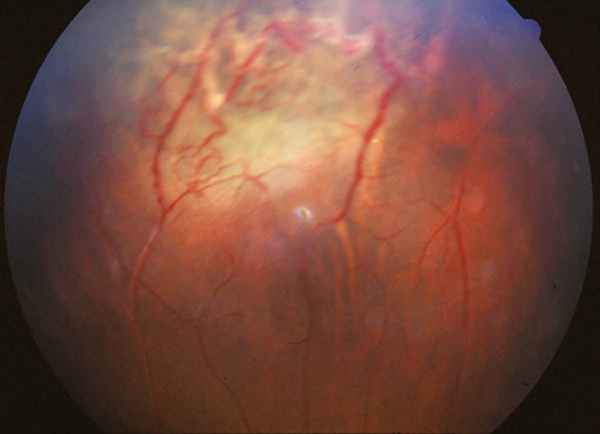
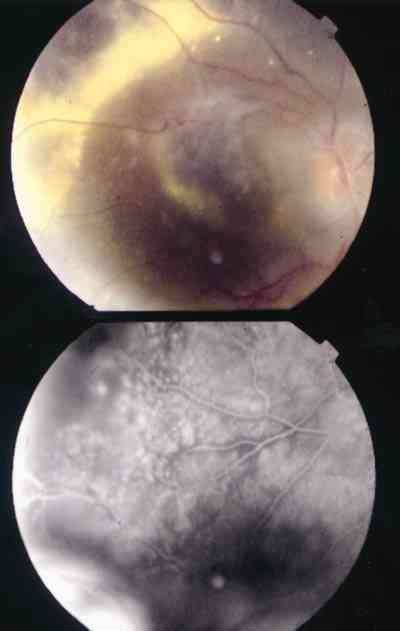
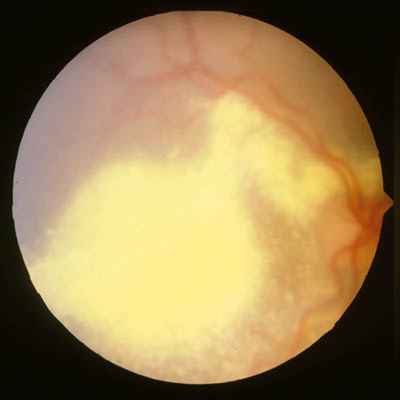
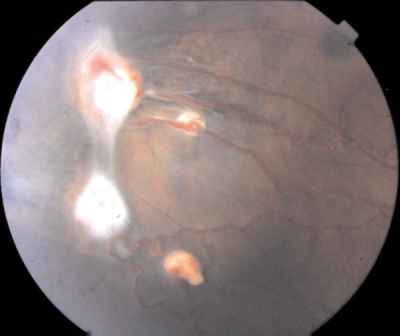
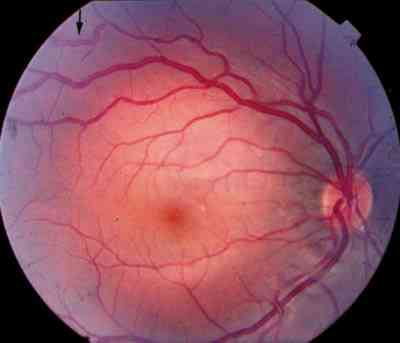
Características clínicas La enf. de Coats tiene una edad media en el diagnóstico de entre 9 y 10 años. Afecta a varones tres veces de forma más frecuente que a mujeres y es unilateral en más del 80% de casos sin haberse observado transmisión hereditaria (11-15). Los síntomas más frecuentes de presentación dependerán de la edad del diagnóstico. En los niños más pequeños se suele presentar en forma de leucocoria, estrabismo o a veces los padres refieren que notan algo raro en la mirada del niño. En niños mayores, las quejas más frecuentes suelen ser disminución de agudeza visual progresiva o incluso ser descubierto en un examen rutinario en el colegio (13-15). El cuadro típico oftalmoscópico es muy variable dependiendo del estadio de la enfermedad. En las formas leves a moderadas se suele observar un desprendimiento exudativo que no suele afectar a más de dos cuadrantes, siendo los más frecuentes los de la zona temporal, acompañados de alteraciones vasculares de aspecto telangiectásicos o aneurismático de la periferia. La exudación suele tener un aspecto blanco amarillento debido a su contenido lipídico y puede ser tanto subretiniana como intrarretininana (figuras 1 y 2). En la evolución se va a producir un desprendimiento exudativo total que a veces puede incluso llegar a colocar la retina detrás del cristalino (figura 3). Esta segunda forma se denomina Coats avanzado (14,16). Las alteraciones vasculares pueden ser mínimas y pasar desapercibidas o por el contrario ser el signo predominante del cuadro (figuras 4 y 5).
Histopatología Las muestras patológicas de ojos con enf. de Coats fueron en el pasado abundantes debido a las enucleaciones por sospecha de tumor intraocular o por desarrollo de glaucomas que llevaban a ojos dolorosos (11,15).
El examen patológico con microscopia óptica y electrónica, revela pérdida de células endoteliales y pericitos, con la consiguiente alteración en la pared del vaso, con dilataciones aneurismáticas y telangiectásiscas y pérdida de la barrera hematorretiniana interna. Secundario a esto se observan la exudación intra y subretiniana de predominio lipídico y con abundante macrófagos llenos de lípidos (figura 6).
Diagnóstico El diagnóstico se basará en las características típicas clínicas de la enf. de Coats, aunque en ocasiones deberemos utilizar pruebas diagnósticas accesorias, sobre todo en niños pequeños para descartar principalmente la existencia de un retinoblastoma, ya que un error en su diagnóstico podría acarrear consecuencias fatales para la vida del niño (14,16,17). Otras patologías que se incluirían en el diagnóstico diferencial serían en general todas las causas de leucocoria como la persistencia de vítreo primario hiperplásico, toxocariasis, hemorragias subretinianas organizadas o angiomas (18,19).
La angiografía fluoresceínica nos será de utilidad en casos de enf. de Coats temprana o moderada, que normalmente será diagnosticada en niños mayores siendo las características angiográficas típicas, anomalías vasculares consistentes en una red capilar anormal con capilares dilatados, presencia de dilataciones aneurismáticas y fusiformes con importante rezume en fases tardías. Las arteriolas afectadas a veces terminan en dilataciones macroaneurismáticas, rodeadas por zonas de cierre capilar atravesadas por shunts arteriovenosos (2,15,20-22) (figura 7).
En caso de enf. de Coats más avanzada con un desprendimiento exudativo importante o si existen dudas razonables en el diagnóstico tenemos una serie de pruebas que podemos realizar para intentar llegar al diagnóstico de certeza: 1. Ecografía y eco doppler. La ecografía es una prueba fácil de realizar, sin riesgo biológico y nos será de utilidad sobre todo para descartar la presencia de calcificaciones subretinianas que inducirían a pensar en un retinoblastoma (17). El eco doppler es capaz de detectar vasos en los tumores no calcificados lo que nos diferenciaría el retinoblastoma que es un tumor vascularizado de la enf. de Coats (23). 2. Tomografía computerizada. Nos será de mucha utilidad también para el diagnóstico diferencial con el retinoblastoma y se caracterizaría por la observación de un desprendimiento de retina con una densidad homogénea en el espacio subretiniano. En caso de Coats más evolucionados las densidades subretinianas pueden ser algo más heterogéneas pero sin llegar a la densidad observada en lesiones calcificadas. Si se realiza un TAC con contraste, no se observará una elevación en las densidades subretinianas y sí acaso se notará un aumento de densidad a nivel de la línea de transición con el vítreo que indica una mayor vascularización a nivel intrarretiniano (18). Con las modernas técnicas de TAC espiral que permite una exploración mucho más rápida, incluso esta prueba puede hacerse con el niño no anestesiado (24). 3. Resonancia magnética nuclear. Al igual que en el TAC la retina está por encima de los límites de resolución espacial de la RMN y el desprendimiento es solo visualizado como diferencia de fluidos de distinta composición y densidad. El material exudativo subretinano compuesto de colesterol, ácidos grasos libres, y proteínas es de baja intensidad en T1 lo que es de esperar dada su similitud con la grasa. El contraste entre la exudación subretiniana y la cavidad vítrea se acentúa en T2 siendo visto el material subretiniano como un área hiperintensa y homogénea (19). 4. Examen citopatológico. En pacientes donde se realiza drenaje de líquido subretiniano el examen citopatológico confirmará el diagnóstico mostrando ausencia de células malignas, así como macrófagos llenos de lípidos (17).
Tratamiento Históricamente la mayoría de ojos con síndrome de Coats avanzado en la infancia, fueron enucleados a causa de su difícil diagnóstico diferencial con el retinoblastoma. En la serie que publica Gómez Morales, 13 de 18 enucleaciones fueron debidas a la posibilidad de tumor maligno (11). Esto se corrobora en otras series en las que un número de ojos fueron enucleados con el diagnóstico incorrecto de retinoblastoma (15,25). En los últimos tiempos el mejor conocimiento del patrón clínico de la enf. de Coats y el uso de pruebas diagnósticas más sensibles, como el TAC (17,18,24), ecografía (17) o resonancia magnética (14,19), han permitido un diagnóstico más correcto y por tanto ha evitado la mayoría de las enucleaciones por este motivo. Otro de los motivos históricos para enuclear estos ojos era el desarrollo en su evolución de un desprendimiento de retina exudativo bulloso total, con posterior progresión a cierre angular y neovascularización secundaria llegando a un ojo doloroso y ciego, aunque esto no ocurre necesariamente en todos los casos (11,13,15,16). El manejo óptimo de la enf. de Coats avanzada con desprendimiento de retina exudativo total, es motivo de controversia. Hoy día, la tendencia, es tratar los ojos con enf. de Coats avanzada, incluso aunque éstos sean ya ciegos, ya que se ha visto, que en muchas ocasiones se consigue conservarlos de forma cosmética aceptable y sin dolor. (14,16,17). No obstante, si un oftalmólogo sospecha que se trata de una enf. de Coats, pero es incapaz de descartar la posibilidad de retinoblastoma de forma concluyente, debe optar por referirlo a alguien con mayor experiencia en el diagnóstico diferencial de retinoblastomas, o proceder a la enucleación. Hasta los años 70 el tratamiento de la enf. de Coats era descorazonador (11). En los años 70 se publicaron algunas series usando fotocoagulación o crioterapia con resultados algo más prometedores (12,13,25). En la actualidad el tratamiento de la enf. de Coats va a depender fundamentalmente de la edad del paciente y del grado de afectación y estadio evolutivo de la enfermedad, teniendo en cuenta sobre todo la presencia o no de desprendimientos exudativos. 1. Crioterapia. Se utiliza sobre todo para lesiones periféricas con o sin exudación. Debe intentarse tratar solo las zonas de telangiectasia. La técnica utilizada será la de congelar-descongelar en dos o tres ciclos. En casos de enfermedad avanzada, se puede unir a drenaje de líquido subretiniano como luego describiremos. La exudación puede aumentar inmediatamente después del tratamiento. Es recomendable tratar un máximo de cuatro horas en cada sesión para evitar en lo posible el aumento del DR exudativo (13,15, 17,26) (figura 8). 2. Fotocoagulación con láser. Muchos pacientes con Coats son demasiado jóvenes para colaborar y permitir el tratamiento en la lámpara de hendidura. Tradicionalmente eran tratados con crioterapia o fotocoagulación con arco de xenon bajo anestesia general. La fotocoagulación con arco de xenon, es técnicamente difícil sobre todo en lesiones periféricas y la crioterapia suele producir un desprendimiento de retina exudativo secundario, que se suele resolver de forma espontánea con el tiempo. Hoy en día, en estos pequeños pacientes tenemos la posibilidad de aplicar el láser, a través del oftalmoscopio indirecto, permitiéndonos una excelente visualización de la periferia retiniana que uniéndolo a la posibilidad de realizar depresión escleral, nos va a permitir tratar de forma fácil incluso las telangiectasias más periféricas. Se debe utilizar láser argón verde, YAG doblado, o cualquier otro que sea bien absorbido por la hemoglobina, utilizando grandes tamaños de spot y larga duración. Nosotros tratamos los vasos telangiectásicos intentando cerrarlos y también la retina isquémica (figura 9). A veces se necesitan varias aplicaciones para cerrar estos vasos. Esta técnica también es usada por algún otro autor (27).
Budning en su serie realiza una barrera de láser en el límite del desprendimiento asociado a crioterapia en las telangiectasias, observando una menor evolución de la exudación hacia el polo posterior (26). En pacientes cooperadores se puede usar el láser a través de la lámpara de hendidura. Se usa spot de 500 micras y tiempos entre 0,2 y 0,5 segundos, intentando producir un espasmo del vaso y quemadura blanca en la retina adyacente (13). 3. Drenaje del líquido subretiniano. Se suele utilizar en casos avanzados con gran componente exudativo, para permitir, un efecto adecuado del frío o del láser. Existen diferentes técnicas dependiendo de los autores. Rydley et al (13), utilizan un cerclaje para restaurar una mejor presión intraocular, así como, relajar tracciones ví treas, mientras que Silodor (16) realiza una esclerotomía postecuatorial y pone una infusión por pars plana o limbo quirúrgico a presión para forzar el drenaje de líquido subretiniano, a la vez, que mantiene una buena tensión en el ojo, todo ello, mediante visualización con oftalmoscopio indirecto, inyectando después aire o gas a presión para mantener la aposición retinocoroidea, pasando entonces a realizar crioterapia de vasos telangiectásicos. Esta técnica ha sido utilizada también por algún otro autor con buenos resultados (14)
En muchas ocasiones, una de estas técnicas o varias de ellas, deben ser repetidas varias veces, hasta el completo cierre de todos los vasos telangiectásicos, la desaparición de la exudación subretiniana, su disminución, o al menos su estabilización, ya que a veces, no se consigue su total desaparición. El número de sesiones en las diferentes series fueron desde una hasta nueve de las distintas terapias (13-17). Como ya se ha dicho, la severidad de la enfermedad, es mayor en niños cuanto más pequeños por lo que éstos suelen requerir mayor número de tratamiento y de forma más agresiva que los mayores (13). En casos no muy avanzados si el tratamiento ha sido efectivo, la obliteración de los vasos, así como, una cicatriz pigmentada suele estar presente en el primer mes y en unos meses se observará una disminución gradual de los exudados, hasta la desaparición, aunque los nódulos sólidos de exudación sobre todo a nivel foveal no desaparecerán sino que irán hacia la fibrosis (13) (figuras 10 y 11).
Se han descrito casos en los que se producen membranas epirretinianas maculares, debidas al tratamiento con láser o crioterapia, que incluso, pueden llegar a producir desprendimientos traccionales en casos de Coats juveniles (28,29). En otros casos se describe la aparición de estas membranas previas al tratamiento (21,22) (figura 7). Estos casos pueden ser una indicación de vitrectomía para liberar dichas tracciones o extraer dichas membranas, aunque se ha descrito la separación espontánea de dichas membranas después del tratamiento de las alteraciones vasculares con láser (22).
Evolución y pronóstico La evolución natural es variable, aunque generalmente progresiva, fases de exacerbación aguda pueden ir seguidas de estabilización durante tiempo. En general sin tratamiento las complicaciones secundarias como iridociclitis, cataratas y glaucoma neovascular o congestivo, pueden llegar a producir ojos dolorosos y finalmente ptisis bulbi (11,13-16), aunque esto no siempre ocurre (17). La enfermedad de Coats sobre todo en niños menores de 4 años, suele evolucionar de forma muy agresiva y es frecuente que el desprendimiento de retina sea total y con gran exudación en el momento del diagnóstico ya que los niños pequeños no se suelen quejar de mala visión en un ojo (13,14,17,26). La mayoría de los trabajos consideran un éxito de tratamiento en estos casos, cuando han conseguido parar la progresión de la enfermedad con disminución importante o desaparición de la exudación subretiniana, aunque en estos casos la recuperación de agudeza visual fue mínima a causa de la gliosis subrretiniana foveal y la ambliopía (13,14,16). Budning en su serie de 25 casos ha encontrado que los factores más importantes que determinan el pronóstico visual en un ojo con enf. de Coats son 1. la cantidad de retina periférica afectada por la telangiectasias, siendo mejor pronóstico una afectación menor de 5 horas así como 2 la no presencia de DR exudativo en el momento del diagnóstico (26).
ANGIOMA CAPILAR DE LA RETINA Y ENFERMEDAD DE VON HIPPEL-LINDAU Los angiomas capilares retinianos son tumores histológicamente benignos que pueden aparecer en la retina periférica o alrededor de la papila. Ambos se pueden dar de forma aislada (30) o ser una de las manifestaciones del síndrome de Von Hippel-Lindau (VHL) (31), siendo idénticos en sus características clínicas, histología y evolución (30).
Síndrome de Von Hippel-Lindau El síndrome de VHL es una enfermedad multisistémica que se hereda de forma autosómica dominante, con expresividad variable, caracterizada por la predisposición a desarrollar una amplia variedad de tumores, siendo los más frecuentes el hemangioblastoma de retina y sistema nervioso central, el carcinoma de células renales, feocromocitoma y quistes renales, pancreáticos y epididimales (31). La incidencia de nacimientos de heterozigotos es de 1:36.000 y la penetrancia es casi completa a los 65 años, aunque la enfermedad puede estar ya presente en la infancia o ser diagnosticada en la séptima década. La esperanza de vida media es de 49 años y el carcinoma de células renales, la causa más común de muerte (31). El aislamiento del gen del síndrome de VHL en el brazo corto del cromosoma 3p en 1993, nos ha dado la oportunidad de determinar las bases genéticas del angioma ocular. El gen se piensa que funciona como un gen supresor tumoral típico, por lo que necesitaría de la inactivación homocigótica de ambos alelos VHL, para iniciar la tumorigénesis en una célula susceptible (32). El síndrome de VHL es un paradigma de la importancia del diagnóstico precoz y el screening. Dado que el angioma retiniano suele ser el primer tumor del VHL en desarrollarse y ser diagnosticado (31), el oftalmólogo juega un importante papel en el diagnóstico precoz de esta enfermedad, ya que el reconocimiento y tratamiento de las manifestaciones del síndrome VHL en un estadio temprano, son importantes para reducir la morbimortalidad, por lo que Webster recomienda el siguiente programa de screning (tabla 1) (30).
Cuando existe historia familiar del síndrome de VHL, la detección de un hemangioblastoma retiniano o del sistema nervioso central es suficiente para su diagnóstico. Si no existe historia familiar son necesarias un mínimo de dos complicaciones para el diagnóstico (ej. dos o más angiomas retinianos o del SNC) o un angioma solitario en asociación con una manifestación visceral (33). Como ya hemos dicho, pueden darse angiomas retinianos esporádicos no asociados a síndrome VHL, aunque son mucho menos frecuentes. Dado que la edad de presentación cuando son sintomáticos, morfología, localización anatómica y complicaciones son similares a las que se ven en el síndrome VHL y teniendo en cuenta que la sensibilidad de los test de ADN para detectar la enfermedad es incompleta, en los casos de angiomas solitarios se recomienda también el mismo screening periódico (30).
Características clínicas En un estudio en el año 98, donde se revisaron los casos de angiomas aparecidos en la literatura entre 1964 y 1998, se vio que la edad media de presentación de angiomas aislados fue de 48 años, mientras que si se asociaban al síndrome de VHL esta edad media era de 25 años (34), aunque en otro estudio posterior se demuestra que cuando los angiomas solitarios eran sintomáticos, la edad de diagnóstico era similar a la de los enfermos con VHL (30). Los angiomas retinianos son raros en edad pediátrica, en la serie de Maher menos del 1% se dieron en niños menores de 5 años y menos del 5% en niños menores de 10 años (31). La edad de diagnóstico, como es lógico, suele ser menor en individuos que se presentan con síntomas teniendo además estos últimos más probabilidades de pérdida de visión. No se ha encontrado predilección por raza o sexo (30). Los síntomas de presentación en edad pediátrica van desde descubrimiento de disminución de agudeza visual en exploración rutinaria del colegio, de forma rara leucocoria, o en caso de niños mayores disminución de agudeza visual o metamorfopsia, dependiendo del grado de afectación macular. En caso de síndrome de VHL el diagnóstico a veces se realiza durante la exploración de screening al existir algún miembro de la familia ya diagnosticado o al ser remitido por otro especialista con diagnóstico previo de VHL (30,35-37). Los hemangiomas capilares pueden desarrollarse en la retina periférica o en la papila y a su alrededor. Los hemangiomas capilares retinianos periféricos, también llamados tumores de Von Hippel pueden tener diferente aspecto según su estado evolutivo. Típicamente son de color rojo o rosado, forma oval y se asocian con un shunt arteriovenoso entre una arteria nutricia muy dilatada y tortuosa y una vena de drenaje (figuras 12 y 13).
El tamaño varía desde microscópicos hasta mayores de 5 mm de diámetro. Los angiomas más pequeños, también llamados microangiomas (37), suelen ser asintomáticos e incluso pueden no ser detectados mediante oftalmoscopia y ser visto solo con angiofluoresceingrafía (31,37). La localización más frecuente es en la retina supratemporal (38). Los angiomas más grandes son los que de forma más común estarán asociados a complicaciones que son las que nos van a producir disminución o pérdida de agudeza visual. En la serie de Webster (38), la complicación más frecuente es el desprendimiento de retina exudativo, seguido de la angiomatosis secundaria en las áreas cercanas a la retina tratada o desprendida, exudación intrarretiniana que puede afectar a la mácula, desarrollo de membranas epirretinianas maculares, hemorragias vítreas, desprendimientos de retina regmatógenos, y neovascularización papilar o retiniana. Los angiomas menores de 0,5 mm o 1/3 del diámetro papilar no suelen causar complicaciones que produzcan disminución de agudeza visual (35,38,39). En casos más avanzados incluso se puede producir proliferación fibrosa sobre la retina y a nivel del vítreo que en su evolución nos van a provocar un desprendimiento de retina traccional con ectopia macular. Esta tracción puede desgarrar la retina llevándonos a un desprendimiento de tipo mixto traccional regmatógeno (29,40). La neovascularización papilar o retiniana, se da en estos pacientes a pesar de tener el resto de la retina y su vascularización sana. Esto se podría explicar, porqué se ha visto una alteración en la regulación del factor de crecimiento endotelial en cultivos de células VHL (41). A veces, sufren regresión espontánea aunque es poco frecuente, apareciendo como lesiones blanquecinas con vasos nutricios muy atenuados, permaneciendo hiperfluorescentes en la AFG, siendo también denominados angiomas fibrosos (37). Los angiomas capilares papilares son más frecuentes que los periféricos si comparamos el área de superficie papilar con la retiniana, aunque en términos absolutos son menos frecuentes (38). Pueden adoptar dos formas clínicas fundamentales: endofítico y exofítico. Los endofíticos se desarrollan a partir de los capilares de los estratos retinianos superficiales y crecen hacia el vítreo como un nódulo rojizo o anaranjado bien delimitado, aunque sin vasos aferentes ni eferentes como el angioma capilar periférico. El exofítico se desarrolla desde los estratos retinianos más externos y suelen tener un crecimiento sesil de bordes mal definidos, produciendo una imagen oftalmoscópica peculiar que es a menudo confundida con papiledema unilateral, papilitis, neovascularización coroidea o hemangioma coroideo (42,43). Las complicaciones asociadas con los angiomas papilares y que llevan a una disminución o pérdida de la visión incluyen exudación intrarretiniana, membranas epirretinianas maculares, estrías o tracción macular, desprendimiento de retina exudativo y hemorragia vítrea (35,38,43).
Histopatología Los estudios histológicos de ojos enucleados describen el angioma retiniano como un tumor compuesto de una proliferación de capilares y células gliales como estroma. Los grandes capilares que forman el tumor muestran su endotelio, membrana basal y pericitos normales, siendo las células estromales identificadas como astrocitos conteniendo grandes vacuolas rellenas de lípidos. En el caso de angiomas yuxtapapilares se ha visto que en ocasiones existen comunicaciones vasculares tanto con la circulación retiniana como coroidea (44,45).
Diagnóstico diferencial El diagnóstico del angioma capilar periférico con sus características clínicas típicas, de masa intrarretiniana rojo anaranjada, con vasos nutricios muy dilatados, es fácil con la oftalmoscopia indirecta y más si existen antecedentes de enfermedad de VHL. La angiografía fluoresceínica es una prueba diagnóstica fundamental sobre todo en los estadios iniciales de su desarrollo, en donde a veces es difícil indentificarlos mediante oftalmoscopia (30,31,37,46) Las características angiográficas típicas son un rápido llenado de la arteria nutricia y del tumor en tiempos tempranos de la AFG demostrando su origen vascular. En tumores pequeños no habrá extravasación de contraste, pero sí existirá fuga de colorante a espacio vítreo en los tumores mayores. También nos será de utilidad para valorar el efecto del tratamiento (46). En edad pediátrica, aunque son raros, es fundamental el diagnóstico diferencial con el retinoblastoma que a veces puede tener vasos nutricios aunque nunca tan dilatados como el angioma, así como el angioma tener una superficie de aspecto blanquecino que puede confundirnos. También se ha descrito algún caso de desprendimiento de retina exudativo, que ha llevado a la enucleación para confirmar el diagnóstico (30). En estos casos puede ser de utilidad pruebas accesorias como ecografía, ecodoppler, TAC o RNM, para realizar un buen diagnóstico diferencial. Otras patologías que nos pueden producir exudación macular o desprendimiento de retina exudativo en esta edad, son el Coats (11), lesiones vasoproliferativas de aspecto tumoral, como las descritas por Shields, aunque ninguno de ellos presenta unos vasos nutricios tan dilatados como el angioma capilar (47). Las malformaciones arteriovenosas congénitas retinianas, como el angioma cirsoide o el hemangioma cavernoso, aunque tienen shunts arteriovenosos dilatados, no presentan la tumoración en el centro del shunt como el angioma retiniano (48). Los angiomas papilares, como ya se ha dicho, pueden ser confundidos con papiledema unilateral, coroiditis peripapilares, o neovascularización coroidea yuxtapapilar, siendo la angiografía fluoresceínica y la clínica acompañante, importantes para el diagnóstico diferencial (43).
Tratamiento Aunque los angiomas retinianos son lesiones benignas, pueden crecer y producir pérdida visual debido a las complicaciones ya comentadas, llegando incluso a la ceguera. Se ha visto gran variabilidad, tanto en la velocidad de crecimiento del tumor, como en la agresividad de la enfermedad, desde la no evolución durante años, hasta un rápido deterioro de la visión (49). Dado que el pronóstico visual es mucho mejor cuando son detectados y tratados en una etapa asintomática, se recomienda su tratamiento nada más sean detectados ya que se ha visto que las complicaciones del tratamiento en estos casos son mínimas (31,37,38,49,50). El tipo de tratamiento va a depender del tamaño, localización y presencia de complicaciones fundamentalmente. Fotocoagulación con láser. Es el tratamiento de elección para la mayoría de angiomas no complicados (33,37,46,49,50). El láser más usado es el argón verde, aunque el YAG doblado tiene un longitud de onda (532 nm), que será mejor absorbida por la hemoglobina y últimamente se está extendiendo su uso. Se utilizan spots de entre 250 y 500 micras, con tiempos de exposición de entre 0,2 y 1 segundo empezando con bajas intensidades hasta producir una quemadura blanca. En tumores pequeños y planos, el tratamiento se puede realizar directamente sobre el tumor, siendo incluso suficiente con una sesión. En tumores mayores de 1 diámetro de papila, se necesitan más sesiones, incluso algunos autores realizan primero un tratamiento sobre la arteria nutricia justo al lado del tumor para disminuir el flujo y permitir un mejor tratamiento posterior del tumor (50,51). En otra serie realizan un tratamiento de la retina adyacente al tumor para intentar evitar un posible desprendimiento de retina ya que esa zona es más susceptible de roturas debido a la posible tracción sobre el angioma (37). Las complicaciones descritas con el láser, no suelen producir disminución de visión y normalmente se reducen a hemorragias peritumorales retinianas o vítreas. Si el tumor se encuentra cerca del área macular se puede producir un edema de la mácula que generalmente se resuelve en semanas. En ocasiones se producen recidivas de los angiomas tratados que deben ser fotocoaguladas de nuevo (37,50). La atenuación de los vasos nutricios y la ausencia de tumor en angiografía fluoresceínica son un indicador del éxito del tratamiento (33,37,50). Crioterapia. En los años 70 diferentes autores recomendaban el tratamiento con crioterapia de angiomas mayores de 1 diámetro de papila, ya que pensaban que el láser no era efectivo en esos angiomas (46,49,52). Posteriormente se han publicado varias series utilizando el láser en angiomas de hasta 4 mm e incluso algunos de ellos con discreta exudación retiniana, con buenos resultados (37,50,51). En general se acepta que la crioterapia es útil para tumores mayores de 3 mm periféricos, personas no colaboradoras como niños pequeños o cuando los medios son opacos (33). Se utiliza una técnica de congelación-descongelación triple, intentando no dar más de 10 ó 12 aplicaciones por sesión y separando las sesiones entre 6 y 8 semanas. Se ha observado como complicación un aumento de la exudación sobre todo en grandes tumores o en aquellos que la tenían previa al tratamiento. En casos de tratamientos agresivos puede llegar incluso a producirse proliferación vítreo retiniana (49). Radioterapia. Se usa generalmente en tumores grandes. Kreusel ha publicado una serie de 25 ojos con angiomas grandes periféricos de 2 diámetros de papila de tamaño medio tratados mediante braquiterapia con Rutenio-106, con buenos resultados en angiomas menores de 5 mm y cuando no existía un desprendimiento de retina exudativo importante previo (36). También se ha utilizado el tratamiento con bomba de protones en estos casos y en casos de angioma papilar (53). Cirugía escleral y vítreo retiniana. Estas técnicas se utilizan en los casos de angiomas complicados. Si el angioma está produciendo un desprendimiento exudativo suficientemente grande como para no permitir otro tratamiento convencional, deberemos realizar el drenaje del líquido subretiniano transescleral, con colocación de un cerclaje y tratamiento del angioma mediante cualquier técnica de las descritas anteriormente (37,49). En los casos en que se desarrolle una membrana epirretiniana macular previa al tratamiento del angioma, la mayoría de los autores abogan por tratar el tumor con láser o crioterapia y esperar un tiempo, ya que en ocasiones se produce un pelado espontáneo de la membrana después del tratamiento, aunque no está claro el tiempo que se debe esperar. Si después de unos meses no se ha producido el pelado espontáneo, la realización de una vitrectomía con extirpación de la membrana suele dar buenos resultados (40,54-56). En los casos más complicados donde existe un desprendimiento traccional o mixto traccional regmatógeno, es necesaria la colocación de un cerclaje para relajar tracciones periféricas, así como realizar vitrectomías más extensas llevando a cabo la resección de las membrana fibrovasculares de forma cuidadosa, con técnicas de cirugía bimanual y utilizando perfluorocarbonos líquidos para manipular la retina en los casos en los que existan agujeros, así como fotocoagulación de éstos, utilizando posteriormente tamponadores de larga duración como C3F8 o aceite de silicona, éste último muy útil en niños por su dificultad para mantener la postura en el postoperatorio. Además se debe realizar tratamiento de la masa tumoral bien con láser endoocular, crioterapia transescleral o endoocular. En las dos series más grandes de vitrectomía en este tipo de patología consultadas, obtienen unos resultados funcionales bastante aceptables. Las complicaciones más frecuentes fueron hemovítreos generalmente limitados, recidivas de membranas epimaculares y desprendimientos de retina tanto regmatógenos como traccionales que necesitaron ser reintervenidos (40,54). El angioma capilar papilar es de manejo mucho más difícil que el periférico por su localización y generalmente tanto su evolución natural como su tratamiento van seguidas de una disminución de agudeza visual sin existir en este momento un tratamiento ideal (35,38). Shields (57) propone, basado en su experiencia, el siguiente algoritmo de tratamiento:
García Arumí propugna la utilización de láser argón verde sobre la superficie del tumor localizado en la papila y de diodo sobre la porción de tumor retiniano, previniendo en parte la lesión del estrato de fibras nerviosas sobre todo en las lesiones en el haz papilo macular. También ha utilizado termoterapia transpupilar aunque su experiencia no ha sido efectiva teniendo que tratar posteriormente con láser (35). Otros tratamientos como diatermia (46), fotocoagulación con arco de xenon (46), resección tumoral externa (58), no son de elección hoy día.
Pronóstico Los factores pronósticos más importantes son la edad al diagnóstico, el tamaño, la localización y la presencia de síntomas en el momento del diagnóstico. En varias series se ha visto que a menor edad en el momento del diagnóstico era mayor la posibilidad de pérdida de agudeza visual (37,38). A menor tamaño del angioma las posibilidades de complicaciones eran menores siendo además el tratamiento mucho más efectivo (37,38). Los angiomas de localización papilar tienen un pronóstico mucho peor por encontrarse sobre el nervio y a veces sobre el haz papilomacular, por lo que tanto su evolución natural como su tratamiento, nos van a provocar pérdida de agudeza visual (35,38). También se ha visto que cuando el diagnóstico se efectuaba por los síntomas, el pronóstico visual era mucho peor, ya que la presencia de síntomas implica la presencia de complicaciones, que aunque sean tratadas con éxito casi siempre van a llevarnos a una disminución o pérdida de agudeza visual (35,37,38). Es muy importante la detección precoz de individuos con síndrome VHL y su familia, al ser una condición autosómica dominante, no sólo porque el pronóstico visual es muy bueno cuando son detectados en fase asintomática, sino también por las posibles repercusiones para la vida del individuo (31). Los pacientes con múltiples angiomas tienen predisposición a la aparición de nuevos angiomas. Debido a ésto, es conveniente vigilar periódicamente a estos pacientes después del tratamiento. Como regla general, Schmidt (37) propone una revisión dos veces al año en pacientes hasta los 30 años ya que se ha visto que el riesgo de pérdida visual es mayor antes de los 30 años (38), y a partir de esa edad una vez al año, o cada dos a tres años, ya que la posibilidad de nuevos angiomas es más rara (31,37,38,46).
BIBLIOGRAFÍA
|