|
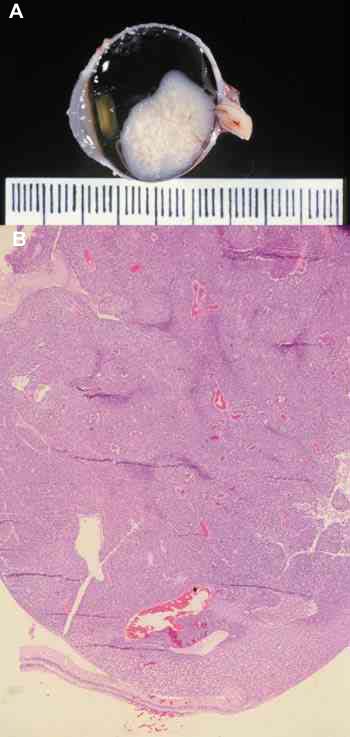
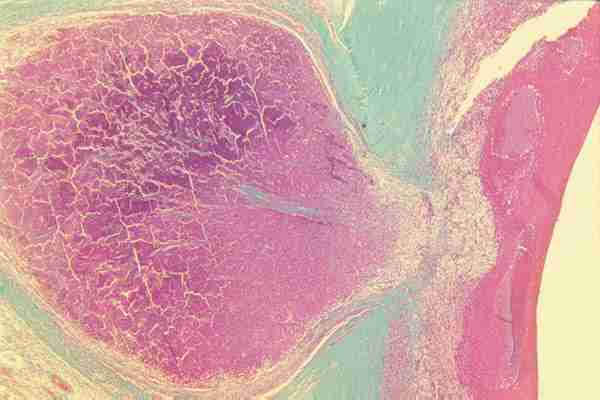
INTRODUCCIÓN El retinoblastoma puede originarse de cualquiera capa de la retina sensorial y en su desarrollo puede presentar diferentes patrones. Retinoblastoma Endofítico: El tumor crece hacia el interior del globo ocular hacia la cavidad vítrea (figuras 1A y 1B) y generalmente la retina no está desprendida.
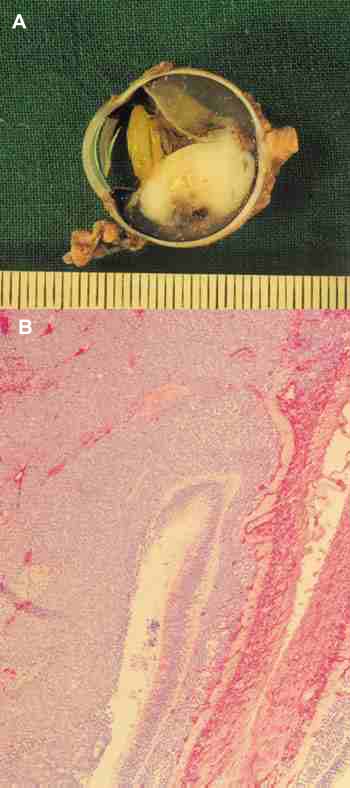
Retinoblastoma Exofítico: el crecimiento se realiza hacia el espacio subretinal y desprende la retina (figuras 2A y 2B).
Retinoblastoma Mixto: la mayoría de los tumores adoptan este patrón, combinanado ambos patrones de crecimiento. Retinoblastoma Infiltrativo difuso: El tumor en su crecimiento produce un engrosamiento difuso de la retina sin que se observen masas tumorales. Frecuentemente sobretodo en un Retinoblastoma endofítico se produce una diseminación vítrea que puede empeorar el pronóstico (figura 3).
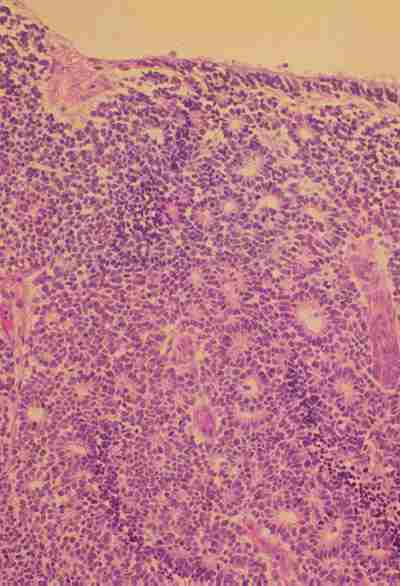
Citológicamente en el retinoblastoma se encuentran 2 tipos de células diferenciadas e indiferenciadas que configuran distintos patrones histológicos que tienen repercusión pronóstica. El Retinoblastoma indiferenciado está compuesto por una proliferación difusa en sábana de células semejantes a las de la retina embrionaria que no muestran evidencia de maduración. Son células pequeñas, redondas o poligonales, con núcleos hipercromáticos, irregulares frecuentemente en mitosis a veces de carácter atípico y escaso citoplasma (figura 4).
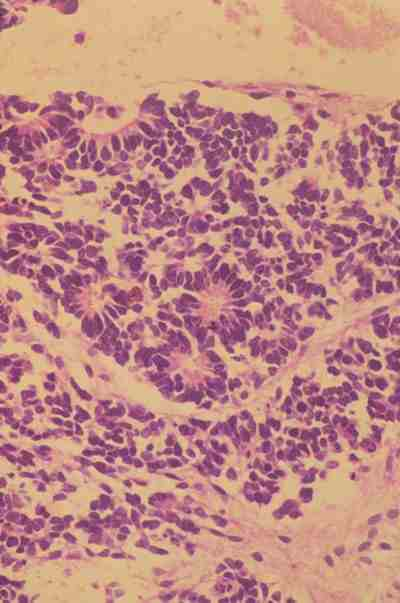
El Retinoblastoma diferenciado es menos frecuente y casi nunca es una forma pura, la diferenciación se traduce no sólo en la formación de rosetas que representan una diferenciación hacia fotorreceptores sino también en la aparición de células semejantes a las células bipolares. Muchos autores admiten que los retinoblastomas diferenciados tienen un mejor pronóstico (1). En las rosetas de Flexner-Wintersteiner se observan células cuboideas o columnares que rodean una luz central que contiene glicosaminoglicanos, delimitada por una membrana eosinófila retráctil, que ultraestructuralmente son barras terminales semejantes a la membrana limitante externa de la retina normal. Los núcleos de las células son basales y se orientan hacia la luz (figura 5).
Menos frecuentemente se forman rosetas desprovistas de luz (rosetas de Homer-Wright), donde las células tumorales rodean una malla fibrilar (figura 6), son menos especificas que las anteriores, pues aparecen en neuroblastomas y meduloepiteliomas.
En algunos Retinoblastomas bien diferenciados se observan un fallo en la diferenciación de fotorreceptores que en lugar de delimitar estructuras circulares, aparecen en una disposición li neal, que se asemeja a un «ramo de flores», «fleurettes», son estructuras eosinófilas compuestas por grupos de células tumorales con prolongaciones eosinófilas en forma de pera que se proyectan hacia una membrana fenestrada (figura 7).
La pseudoroseta es un término confuso que no tiene valor pronóstico. Las células presentan una disposición semejante a las rosetas, y pueden corresponder a:
Otro tipo peculiar de diferenciación célular descrita más recientemente (1) es una célula de tamaño un poco mas grande, de núcleo pequeño redondo y central, con citoplasma amplio y poligonal, que se asemeja a la célula bipolar de la retina normal y se dispone en grupos o nidos. Estas células como los rosetas de Flexner-Wintersteiner indicarían una marcada diferenciación (figura 8).
Los retinoblastomas diferenciados, suelen expresar positividad para la sinaftofisina (figura 9).
La mayoría de los tumores muestran amplias áreas de necrosis, causadas por isquemia o respuestas inmunológicas, que frecuentemente se acompañan de calcificaciones (figura 10).
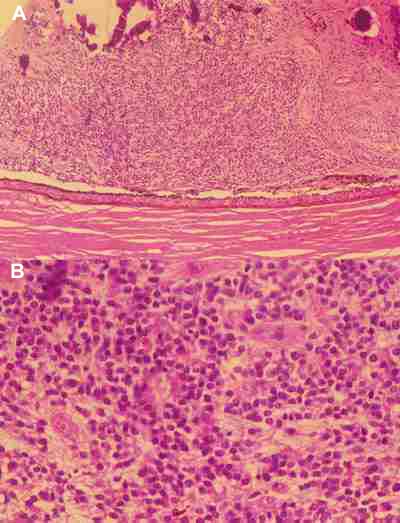
El retinoblastoma puede extenderse a lo largo del nervio óptico, hacia el cerebro, o las células neoplásicas pueden penetrar a través del espacio subaracnoideo que rodea al nervio. La infiltración neural puede ser pre-laminar, laminar o post-laminar. El riesgo de metástasis es elevado si la invasión del nervio es post-laminar (figura 11) (2,3).
También puede producirse una diseminación extra-ocular a través de los canales esclerales que contienen vasos y nervios. La invasión focal de la coroides no parece modificar el pronóstico si la invasión es masiva lo empeora (4) (figura 12).
MATERIAL Y MÉTODO Se estudian 13 casos de retinoblastoma (dos de ellos pertenecen a un mismo paciente), tratados con quimioterapia y terapias asociadas, que fueron enucleados por complicaciones derivadas del tratamiento. En todos los casos se estudió el porcentaje de tumor viable, después del tratamiento y en el caso de persistencia tumoral el grado de diferenciación. Se valoró la intensidad de la necrosis, la presencia de calcificaciones y la existencia de fibrosis que sustituye al tejido tumoral. Asimismo también se estudió la proliferación glial acompañante y la presencia de histiocitos en los focos de fibrosis. En los tumores indiferenciados se valoró el grado de atipia citológica y el número de las mitosis y en los tumores diferenciados las rosetas de Flexner Winterteiner, las rosetas de Homer-Wright, las «fleuretes», la diferenciación bipolar y la existencia de pseudo-rosetas. En cada caso se estudió la proporción de diferenciación o indiferenciación tumoral y algunos fueron seleccionados para estudios inmunohistoquímicos de: sinaptofisisna (SNS), enolasa neuronal específica (NSE), S100, y proteina glio-fibrilar (GFAP). En todos los casos se estudió el índice de proliferación celular utilizando el anticuerpo MIB-1 que reacciona con el antígeno nuclear Ki-67. Se estudió el nervio óptico para valorar la existencia de infiltración preliminar, laminar o post-laminar y también fueron revisadas las envolturas oculares para localizar infiltraciones tumorales. Los ojos enucleados mostraron diferentes patrones de regresión y fueron agrupados según el siguiente esquema (tabla 1).
Regresión total: No existe tumor viable en la masa tumoral. Pueden existir tumores múltiples microscópicos a través de la retina que fueron etiquetados como Regresión tipo X. En casos negativos se tipifica la lesión como Regresión Tipo 0. Regresión parcial: Existe tumor viable en una proporción del 10-20%, la regresión se acompaña de fibrosis, calcificaciones, proliferación glial y presencia focal de histiocitos en algunos casos, la necrosis es mínima o no existe.
Regresión mínima: El tumor persiste en mayor proporción y se acompaña de abundante necrosis y calcificaciones, no existe o es mínima la fibrosis, la proliferación glial y la presencia de histiocitos. El tumor puede ser diferenciado (D), indiferenciado (ID), con tumores múltiples microscópicos (X) o sin éstos (O). Regresión combinada: Se combinan diferentes patrones bien en la misma masa tumoral o en distintas proliferaciones tumorales.
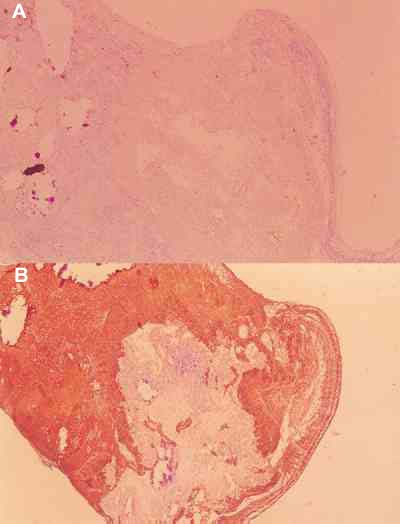
RESULTADOS La regresión total, con ausencia de tumor viable, sólo se observó en un caso (caso n.º 9). La regresión fue total pero se observaron tumores multicéntricos en el resto de la retina que no respondieron al tratamiento y que no mostraron rasgos de diferenciación, observándose a este nivel un elevado índice de proliferación celular con el Ki-67. Por el contrario la masa tumoral fue absolutamente negativa con el Ki-67 y marcadamente positiva con la proteina gliofibrilar (figuras 13, 14A, 14B y 15).
En 5 retinoblastomas se observó regresión parcial. El tumor persistió en una proporción del 10-20% y mostró signos de diferenciación en 4 de ellos (casos 2, 3, 4, 11) (figuras 16A, 16B y 17); en los 3 primeros se observaron crecimientos múlticentricos microscópicos. Sólo en un caso (caso 5), el tumor fue totalmente indiferenciado y también presentó un patrón de crecimiento multicéntrico.
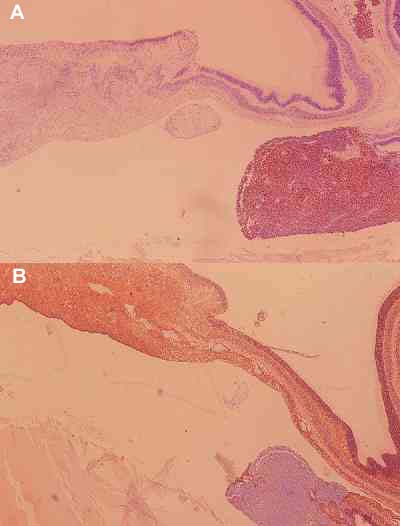
Cinco casos respondieron al tratamiento con predominio de la necrosis, en algunos existían calcificaciones, pero la fibrosis y otras respuestas acompañantes eran mínimas. Este tipo de regresión es el que hemos llamado mínima, en la que existe tumor viable, en mas del 20%. En todos el tumor es indiferenciado y presenta un marcado índice de proliferación celular con el Ki-67 (figuras 18 y 19). Dos de los casos (casos 10 y 13), mostraron un crecimiento masivo, ocupando el tumor la totalidad de la cámara vítrea, y en ambos casos infiltraban el nervio óptico, pre-laminar en el caso 10 y post-laminar en el caso 13, en éste último existía infiltración coroidea. En los 3 casos restantes (casos 1, 7 y 12) el crecimiento no era masivo, pero existía tumor viable en porcentaje elevado y tumores microscópicos multicéntricos que tampoco respondiera al tratamiento, y uno de los casos (caso n.º 7) existía infiltración prelaminar del nervio óptico e infiltración escleral.
En dos casos se encontró el tipo de regresión que hemos llamado combinada, se trataba de casos que mostraban los patrones descritos anteriormente. En el caso n.º 6 existían dos masas tumorales (figura 20), una de ellas no respondió al tratamiento, sólo se observó necrosis, la otra presentó una regresión total con negatividad del Ki 67 (figuras 21A y 21B). Por tanto se combinó un patrón de regresión mínima con otro de regresión total. En el caso n.º 8 se combinó una regresión parcial del tipo mínima. En ambas masas tumorales el tumor persistio indiferenciado y existían tumores multicéntricos múltiples (figura 22).
La tabla 2 adjunta muestra los resultados globales de los casos revisados.
DISCUSIÓN Los patrones de regresión clínicos tras el tratamiento con radioterapia están ampliamente descritos en la literatura, tanto con la braquiterapia en placas epiesclerales como en la radioterapia externa. Por el contrario, los resultados histológicos obtenidos tras el tratamiento quimioterápico son escasos, y salvo el trabajo de Bechrakis (5), no han sido publicados. Creemos que el estudio de series más amplias, la valoración del tumor viable mediante marcadores de proliferación celular y la utilización de tratamientos locales para tumores residuales, pueden contribuir al avance del tratamiento del retinoblastoma.
BIBLIOGRAFÍA
|