|
La órbita posee un armazón óseo formado por los huesos craneales, faciales y nasales. En el borde anterior de la órbita el periostio desarrolla una lámina de tejido conjuntivo denso (el septum orbitario) que se extiende hasta la inserción de los párpados, su parte posterior forma parte también de la órbita. En los huesos que la configuran existen canales, a través de los cuales penetran vasos y nervios en el interior de la órbita. El interior está ocupado por el globo ocular, el nervio óptico con su recubrimiento meningeo, la cápsula de Tennon, los músculos extraoculares, tractos nerviosos que los inervan, la glándula lagrimal, vasos sanguíneos y tejido fibroadiposo que sirve de soporte a estas estructuras. En los tejidos blandos orbitarios también podemos encontrar fascículos de fibras musculares lisas y una pequeña estructura cartilaginosa, la troclea que envuelve fascículos de fibras musculares estriadas del músculo oblicuo superior. En la órbita podemos encontrar lesiones pseudotumorales y auténticos tumores derivados de los tejidos blandos orbitarios, de las estructuras óseas, del nervio óptico y también tumores secundarios originados en las estructuras vecinas que por extensión o metástasis infiltran la órbita. Las anomalías en el desarrollo de los elementos que componen la órbita dan lugar a un alto porcentaje de lesiones pseudotumorales orbitarias. La presencia de canales linfáticos en la órbita está en discusión y en la actualidad se admite que los linfangiomas traducen alteraciones del sistema venoso orbitario por lo que se debería sustituir el término de linfangioma por el de anomalía orbitaria venosa (1). En la órbita no existe tejido linfoide. A partir del infiltrado linfoide que generalmente existe en la lagrimal es de donde se originan los linfomas y pseudolinfomas. La propia glándula lagrimal puede ser causa de procesos neoplásicos (excepcionales en la infancia), por el contrario anomalías en su desarrolllo, crecimiento ectópico o hiperplasias pueden observarse en esta edad.
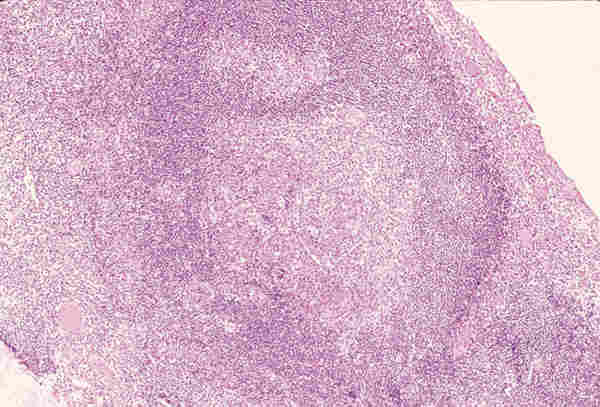
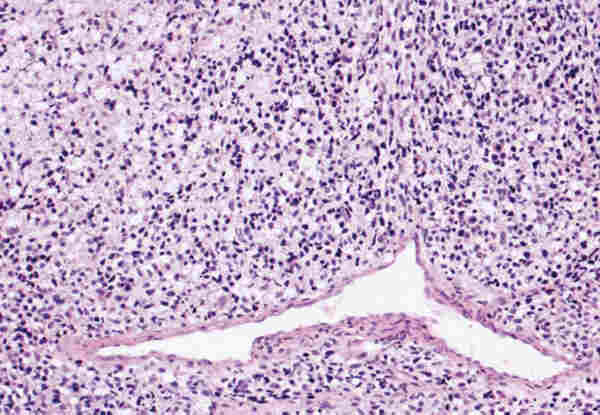
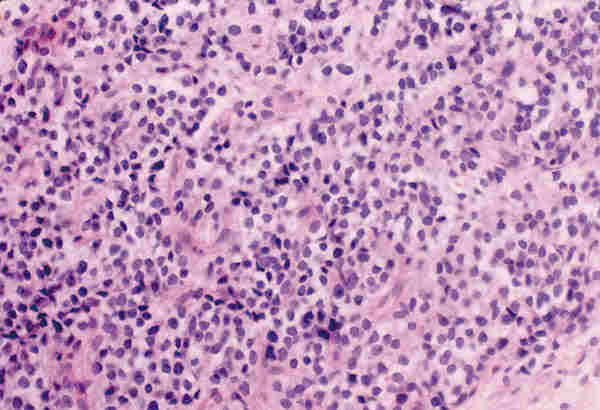
LESIONES PSEUDOTUMORALES Las causas más frecuentes de lesiones pseudotumorales son los procesos inflamatorios que producen celulitis orbitaria que generalmente son secundarias a sinusitis paranasales, procesos inflamatorios de dientes o del saco lagrimal. Estas celulitis son la causa más frecuente de la proptosis en la infancia. El patólogo no suele tener biopsia de esta clase de patología, pues el diagnóstico es fundamentalmente clínico. Su etiología puede ser bacteriana, micótica o parasitaria, siendo estas últimas muy infrecuentes y suelen originar problemas de diagnóstico diferencial con auténticos tumores. El quiste hidatídico en nuestro país puede plantear esta problemática. El término pseudotumor inflamatorio de la órbita es ambiguo (2) y contribuye a la confusión en el manejo y diagnóstico de los enfermos. Durante muchos años bajo este término se han incluido diferentes entidades y se han clasificado según su localización anatómica; miositis (músculo estriado), dacriocistitis (glándula lagrimal), periescleritis (cápsula de Tenon), perineuritis (nervio); según el cuadro clínico (aguda, subaguda y crónica), por su patrón radiográfico (infiltrativo o no infiltrativo) y por su etiología (traumática, inflamatoria, neoplásica, vascular, endocrina o inmunológica). Sólo una vez que se han excluido factores etiológicos locales o sistémicos podremos hablar de un pseudotumor inflamatorio idiopático cuya base anatomopatológica es una inflamación crónica no granulomatosa. Las técnicas modernas de biología molecular y la tipificación celular mediante la inmunohistoquímica nos permiten excluir las lesiones linfoproliferativas monoclonales que más dificultad presentan a la hora de hacer un diagnóstico diferencial. El pseudotumor inflamatorio en patología pediátrica presenta una frecuencia que oscila entre el 8% y el 16% (3-5). La mayoría de estas lesiones tienen como substrato un infiltrado inflamatorio crónico inespecífico compuesto por células plasmáticas, linfocitos, macrófagos y ocasionales eosinófilos, sobre un estroma fibroso denso en el que se puede observar ocasionales folículos linfoides (figura 1).
Existen variantes histológicas del pseudotumor inflamarorio:
Las anomalías en el desarrollo de las estructuras orbitarias configuran otro grupo de lesiones pseudotumorales en la infancia. Son congénitas y se caracterizan por alteraciones en el tamaño, localización, organización o proporción de los tejidos presentes en la órbita (tabla 1) clasificándose en:
1. Coristomas: Es el crecimiento excesivo de un tejido histológicamente normal que durante el desarrollo se desplaza hacia una región anatómica en la que normalmente no existe este tejido. Los coristomas orbitarios se originan de células embrionarias que quedan atrapadas en situación anómala por un fallo en la separación en los tejidos óseos o por un secuestro. Suelen ser quísticos y contienen células que derivan de una sola capa germinal. Se clasifican en:
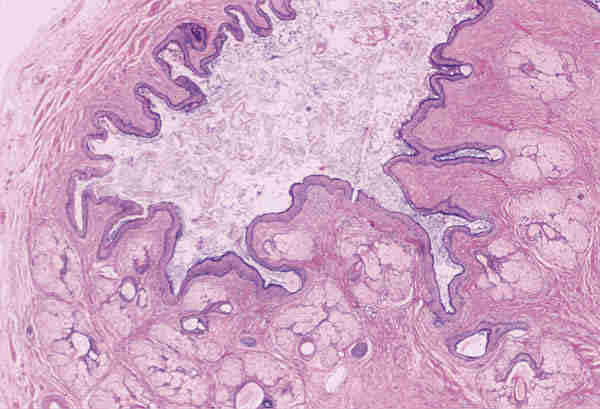
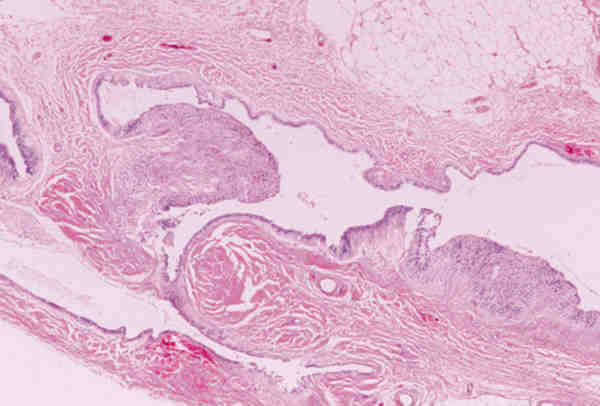
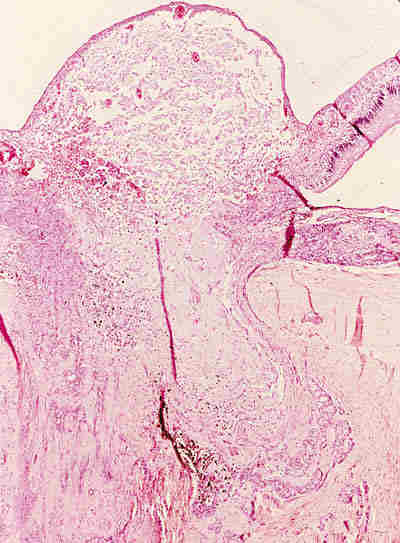
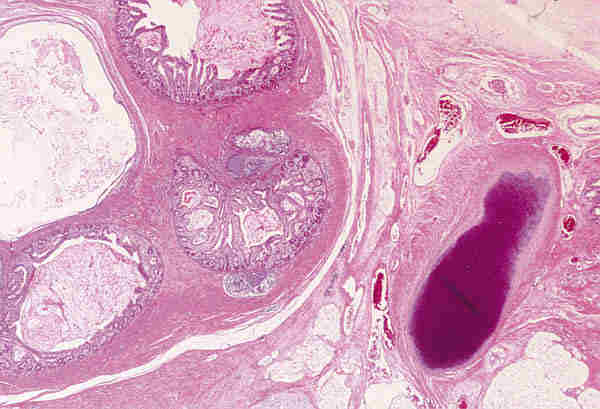
Ambas lesiones suelen aparecer en los primeros meses de vida en forma de nódulos subcutáneos situados sobre el anillo orbitario, aunque a veces también existen en zonas más profundas de la órbita. Son lesiones benignas que no infiltran las estructuras adyacentes y se tratan con extirpación quirúrgica simple. Lesiones quísticas idénticas pueden desarrollarse secundariamente por implantación traumáticas o yatrogénica. Los quiste dermoides conjuntivales (6,7) se originan por un secuestro de la conjuntiva durante la vida embrionaria. Se encuentran revestidos por un epitelio escamoso no queratinizante con células caliciformes y anejos en su pared (figura 4). Si no poseen anejos se denomina quistes simples que suelen ser secundarios y aparecer meses o décadas después del trauma o cirugía.
* Dermolipomas: Son dermoides sólidos compuestos por tejido adiposo, epitelio estratificado y anejos epidérmicos. Son coristomas congénitos que generalmente aparecen en el cuadrante superior temporal y pueden extenderse posteriormente hacia la órbita. Son frecuentes en el síndrome de Goldenhar.
2. Hamartomas: Es el crecimiento excesivo de un tejido histológicamente normal en su localización habitual. Los hemangiomas para muchos autores son un ejemplo de hamartoma.
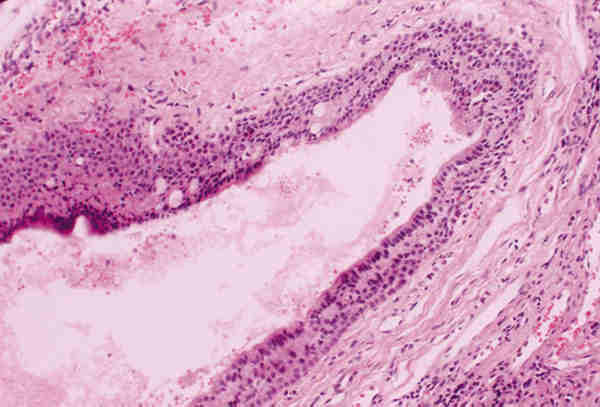
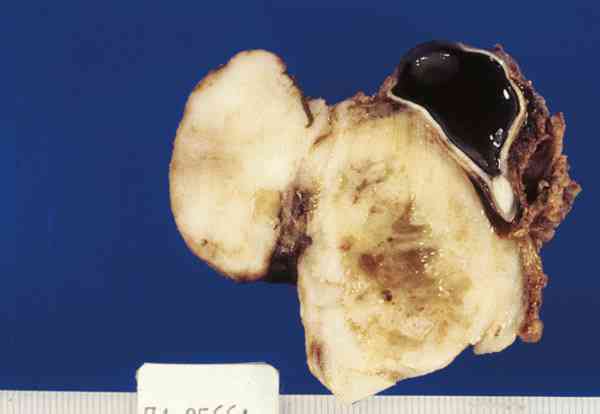
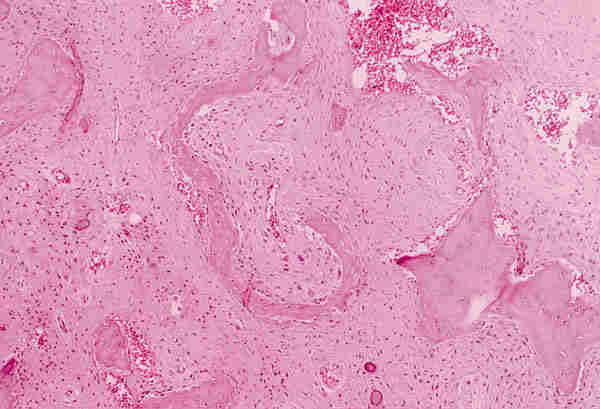
3. Microftalmia con quiste: Es una lesión malformativa, poco frecuente, producida por un cierre incompleto de la hendidura colobómica. A través de este defecto protuye el tejido retino-glial y se desarrolla el quiste. A veces es tan grande la lesión quística que el globo ocular microftálmico es difícil de encontrar y plantea el diagnóstico diferencial con un tumor de partes blandas. Histológicamente el globo ocular puede estar bien o mal organizado y presentar lesiones intraoculares secundarias como displasia retiniana o aparición de otros tejidos (adiposo, óseo o cartilaginoso). El quiste puede estar delimitado por retina gliótica o puede estar totalmente ocupado por una proliferación glial que simula una neoplasia y plantea el diagnóstico diferencial con un astrocitoma (figura 5).
La mayoría de los casos son unilaterales y esporádicos pero se están empezando a conocer casos asociados a anomalías genéticas (trisomía parcial del cromosoma 22) (8) y síndromes sistémicos. En la actualidad (9) según su etiología se clasifican en Genéticos (monogénico y cromosómico), adquiridos prenatales (agentes teratógenos y malformativos intrauterinos) y sindrómicos (asociados a otras malformaciones)
4. Cefaloceles: En este tipo de malformación se produce una herniación de las meninges (meningocele), encéfalo (encefalocele) o de ambos (meningoencefalocele) hacia la órbita, con cierre posterior del orificio de comunicación.
5. Anomalías de la glándula lagrimal: La presencia de una glándula lagrimal ectópica o su crecimiento hiperplásico también puede producir una lesión pseudotumoral en la órbita.
Lesiones tumorales orbitarias Las neoplasias orbitarias pueden ser: A partir de las diferentes estructuras de la órbita se desarrollan tumores que suelen ser benignos y en un 90% de naturaleza vascular. A la cabeza de los tumores malignos primarios de la órbita (tabla 2) se encuentran los rabdomiosarcomas.
b) Secundarias: Se desarrollan a partir de las estructuras vecinas e infiltran la órbita o bien son de naturaleza metastásica (tabla 3).
TUMORES ORBITARIOS PRIMARIOS 1. T. vasculares Las lesiones vasculares de la órbita son difíciles de clasificar pues cumplen criterios tanto de neoplasias (proliferación celular en los hemangiomas) como de hamartoma (con predominio en la infancia y capacidad de regresión). En los hemangiomas infantiles se han descrito tres fases de crecimiento (10), una fase proliferativa, que dura aproximadamente un año, seguida de una fase involutiva que oscila entre 1 y 5 años, al cabo de 6-12 años se puede encontrar totalmente involucionado el hemangioma. Otro problema de su clasificación es la confusión que existe entre malformación vascular y neoplasia vascular. Mulliken (11) diferencia los hemangiomas de las malformaciones vasculares en base a que estas últimas son la consecuencia de una alteración en la morfogénesis vascular durante la vida embrionaria, y por tanto son lesiones congénitas, que pueden aumentar de tamaño por fenómenos ectásicos secundarios a cambios de presión en el flujo vascular o linfático pero que nunca se acompañan de proliferación de células endoteliales a diferencia de los hemangiomas. Por último, como ya hemos mencionado, los hemangiomas para algunos autores (1) representan una anomalía del sistema venoso orbitario que junto a las varices deberían ser consideradas como malformaciones venosas. Otros autores no están de acuerdo y prefieren mantener los dos términos (2,12). Recientemente (13) los miembros de la Sociedad Orbitaria Internacional han adoptado una clasificación de las malformaciones vasculares orbitarias basadas en sus repercusiones hemodinámicas. Esta clasificación no se aplica en aquellas lesiones que producen expansión por proliferación celular (H. capilar, H. cavernosos y neoplasias vasculares de células fusiformes).
Hemangioma capilar Es el tumor vascular más frecuente en la infancia. Puede observarse en el nacimiento o más tarde y frecuentemente afecta a la porción cutánea de los párpados lo que hace más fácil el diagnóstico que cuando se localizan en las porciones superficiales o profundas de la órbita. La imagen histológica (figura 6) corresponde a numerosos capilares con proliferación de células endoteliales. Al involucionar la proliferación capilar es sustituida por tejido adiposo. El tratamiento está en función del tamaño y recientemente se está utilizando el interferón para inhibir la angiogénesis en aquellos casos que no han respondido a los corticoides (14).
Hemangioma cavernoso Histológicamente la lesión corresponde a vasos dilatados ectásicos, revestidos por un endotelio aplanado y rodeado por una gruesa cápsula de tejido conectivo fibroso. Su etiopatogenia está en discusión, para algunos equivale a una malformación vascular con aspecto esponjoso, para otros sería un hemangioma en fase involutiva que adopta esta morfología. Este tipo de hemangioma es la lesión más frecuente en la edad adulta.
Hemangiopericitoma Es un tumor vascular de partes blandas que a veces afecta de forma primaria o secundaria a la órbita. Se localiza más frecuentemente en la mitad superior de la órbita y es infrecuente en la edad infantil. El patrón básico es una proliferación de espacios vasculares rodeados de una proliferación de células fusiformes (pericitos). Su comportamiento es difícil de precisar y no existen criterios histológicos para definir su benignidad o malignidad (15).
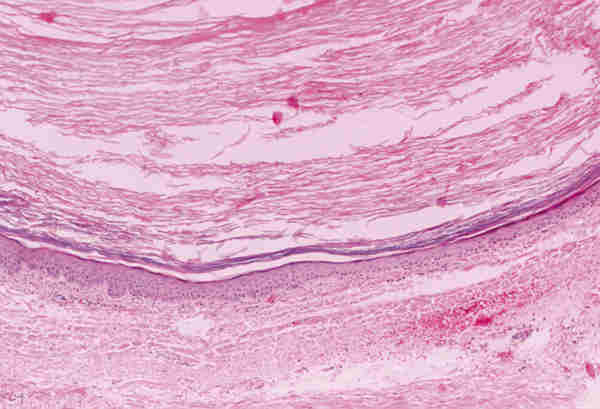
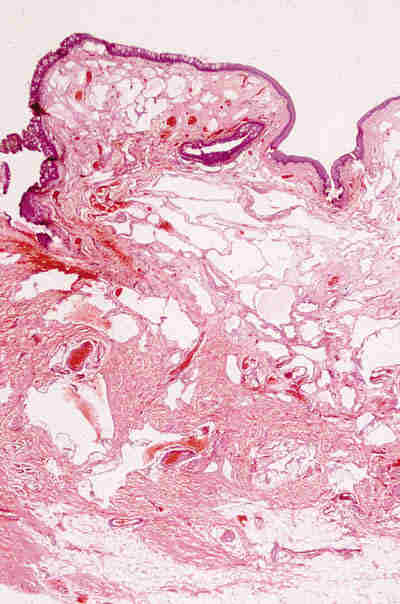
Malformaciones vasculares * Malformación linfática-venosa: La mayoría de las lesiones son visibles desde el nacimiento o aparecen en los primeros años de vida y pueden sufrir un aumento del tamaño por fenómenos hemorrágicos. Se tratan de lesiones desprovistas de cápsula y compuestas por canales de diferentes diámetros lo que les da un aspecto esponjoso. Los canales presentan paredes finas revestidas por endotelio, que contienen sangre o líquido seroso. Se encuentran incluidos en un estroma fibroso y a veces se observan infiltrados linfocitarios focales (figura 7).
* Varices orbitarias: Es una malformación venosa en la que se produce la dilatación de una o varias venas que conduce a una proptosis intermitente en función del aumento de la presión venosa.
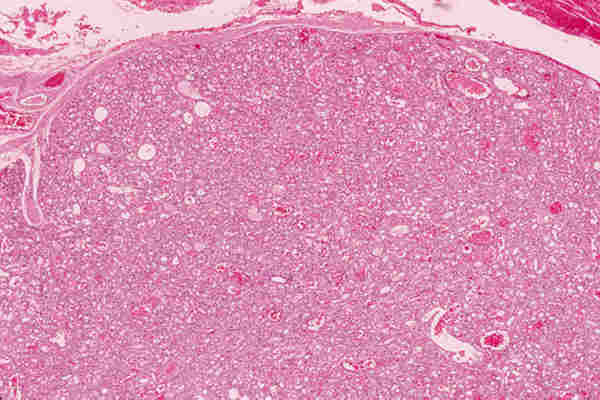
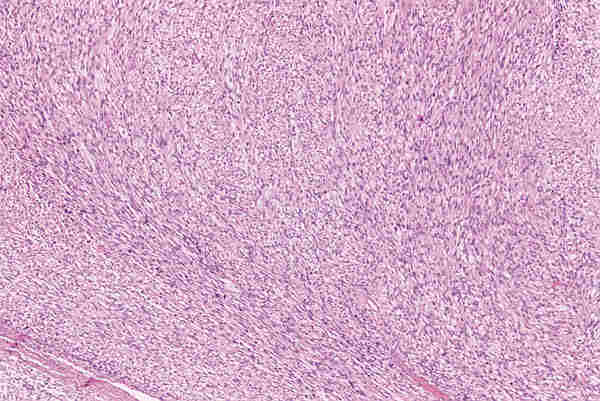
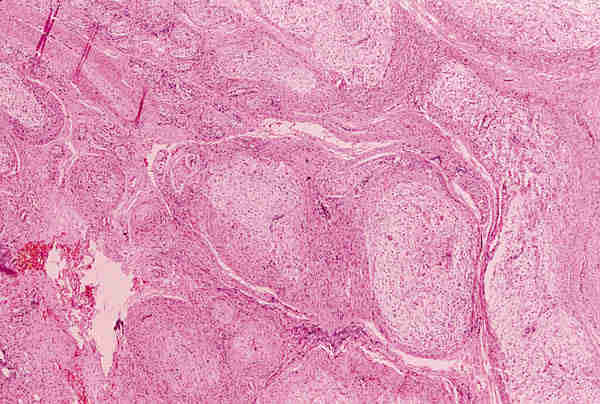
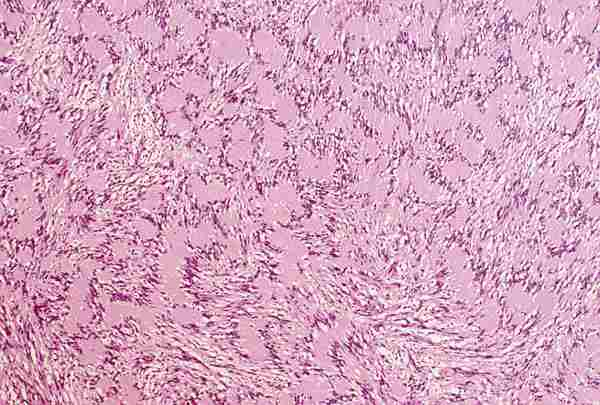
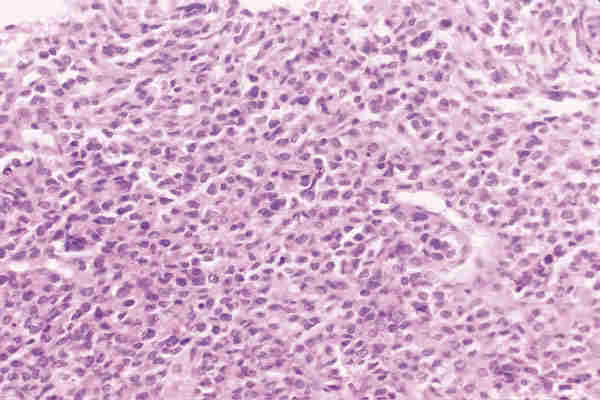
2. T. musculares Los tumores con diferenciación de músculo liso, tanto benignos (leiomiomas) como malignos (leiomiosarcoma) son infrecuentes y aparecen en la edad adulta. Los tumores con diferenciación hacia músculo estriado de comportamiento benigno; rabdomiomas son poco frecuentes (16). Los rabdomiosarcoma, a pesar de su rareza, son los tumores orbitarios malignos primarios más frecuentes en los niños y el sarcoma más frecuente de partes blandas en la infancia (17-19). La edad más frecuente de aparición es entre los 7 y 8 años y el 90% de los casos aparecen antes de los 16 años. Se trata de un tumor de localización unilateral, de crecimiento rápido que produce proptosis y desplazamiento del globo ocular. La introducción de la quimioterapia y la radioterapia como tratamiento coadyuvante a la cirugía ha mejorado llamativamente el pronóstico que anteriormente era infausto (20). De los patrones histológicos descritos en el rabdomiosarcoma por Horn y Enterline (21) en 1958, embrionario-alveolar-botrioide-pleomórfico, son preferentemente los dos primeros los que aparecen en la órbita. Actualmente se utiliza una nueva clasificación realizada por el instituto nacional del cáncer (19) clasificándose en rabdomiosarcoma embrionario, en el que se identifican cuatro variantes —convencional, convencional con áreas de citología agresiva, leiomiomatoso y convencional con areas pleomórficas—, rabdomiosarcoma alveolar; con dos variantes —convencional y sólido—; R pleomórfico y una última categoría correspondiente a otros sarcomas indiferenciados. El R. embrionario, con todas sus variantes, constituye el tipo más frecuente. Está formado por células musculares embrionarias, rabdomioblastos, en diferentes estadios de diferenciación. Podemos observar células redondeadas indiferenciadas y células maduras de morfología fusiforme, citoplasma abundante e intensamente eosinófilo donde a veces se pueden reconocer estriaciones transversales. Estas células se encuentran inmersas en un estroma que con frecuencia tiene características mixoides (figuras 8 y 9). Es importante destacar la variante leiomiomatosa (figura 10) por ser la que mejor pronóstico presenta. El R. alveolar convencional es de peor pronóstico y se caracteriza por la presencia de septos fibroso que delimitan espacios alveolares donde se encuentran «flotando» las células tumorales que son rabdomioblastos totalmente indiferenciados. Aquí se observa un mayor grado de pleomorfismo, con presencia de células gigantes multinucleadas, y un mayor índice mitótico, lo que traduce su mayor agresividad (figura 11). Existe una variante de nueva aparición, Alveolar de patrón sólido (19), que presenta las mismas características citológicas con ausencia del patrón morfológico alveolar y que tiene el mismo mal pronóstico (18) (figura 12).
3. T. adiposos Los tumores benignos, lipomas, a pesar de su rareza representan el 2% de algunas series de tumores orbiculares pediátricos (22). Se encuentran bien delimitados, con una apariencia lobular, y se encuentran constituidos por tejido adiposo adulto, bien diferenciado. Es difícil establecer el diagnóstico diferencial con los prolapsos de la grasa orbitaria (figura 13).
Los malignos, liposarcomas, son raros y los pocos casos descritos aparecen en la edad adulta.
4. T. fibroblásticos o fibrohistiocitarios Los tumores fibrohistiocitarios benignos y malignos (fibrohistiocitomas), y los que presentan diferenciación fibroblástica, existen en la órbita de individuos adultos pero son excepcionales en la edad infantil (23,24).
5. T. con diferenciación neural Constituyen el 4% de los tumores orbitarios (25). El 2% se corresponde a neurofibromas y el otro 2% a neurinomas, siendo excepcionales los neurofibrosarcomas.
* Neurofibroma: Es el tumor más frecuente de las vainas nerviosas periféricas de la órbita. Existen tres patrones de crecimiento: plexiforme, difuso y localizado (o circunscrito). El primero es el que aparece en la edad pediátrica, frecuentemente afecta los párpados y es patognomónico de las neurofibromatosis (25). Histológicamente no están encapsulados y presentan un aspecto organoide, observándose una proliferación de células de Schwann, fibroblásticas y perineurales que engloban los axones (figura 14).
El tratamiento es quirúrgico y algunos autores recomiendan un tratamiento agresivo desde el principio para evitar múltiples intervenciones (26).
* Neurinoma: Clínicamente son indistinguibles de los neurofibromas solitarios de los que se diferencian por encontrarse encapsulados y por estar constituidos por proliferaciones de células de Schwann que adoptan un patrón sólido en empalizada (Antoni A) o un patrón mixoide (Antoni B) (figura 15).
* Neurofibrosarcoma: Son los tumores malignos de las vainas periféricas y son infrecuentes en niños.
6. T. del n. óptico Pueden localizarse a nivel de la cabeza (melanocitoma, meduloepitelioma y hemangioma) o en la porción retrobulbar (glioma y meningioma).
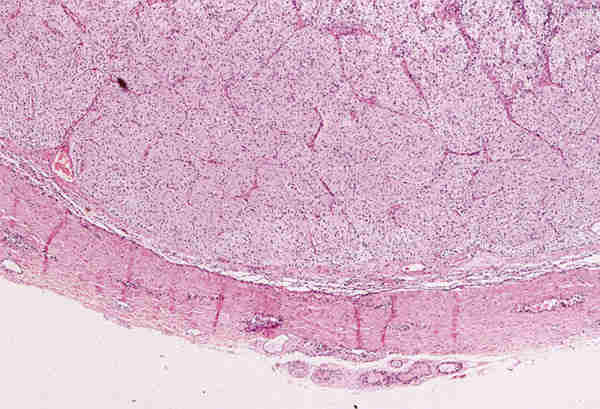
* Gliomas: Aparecen en la primera década, alrededor de los 5 años. Probablemente la mayoría son congénitos y sólo se diagnostican cuando dan síntomas. Son astrocitomas de bajo grado de malignidad de crecimiento lento. Se originan a partir de astrocitos fusiformes (pilocíticos) que proliferan y expanden los septos piales que permanecen intactos (figura 16). No suelen existir atipias citológicas ni mitosis y es frecuente la aparición de fibras de Rosenthal. Se suelen acompañar de una proliferación reactiva, que a veces es tan intensa que plantea el diagnóstico diferencial con un meningioma (27).
Un alto porcentaje de casos aparece igual que el meningioma en enfermos con neurofibromatosis.
* Meningiomas: Los meningiomas primarios orbitarios derivan de células mesodérmicas que quedan incluidas dentro del espacio subaracnoideo del nervio óptico. Adoptan idénticos patrones que los intracraneales (fibroblástico, angioblástico, meningotelial o transicional) existiendo un predominio del meningotelial con aparición de cuerpos de psamoma y disposición celular en capas concéntricas (figura 17).
Son tumores fundamentalmente de adultos pero el 20% de los casos aparecen antes de los 10 años (28). Son de crecimiento lento, localmente invasivos pero no metastatizantes. En niños son más agresivos que en los adultos (29) y la supervivencia es más larga en los meningiomas primarios que en los secundarios (extensión a órbita de un meningioma intracraneal).
* Meduloepitelioma: Es un tumor que aparece fundamentalmente en la primera década (30) y que se origina de células que recuerdan al epitelio medular que reviste el tubo neural. Se localizan preferentemente en cuerpo ciliar pero también se han descrito a nivel del nervio óptico (31). Histológicamente posee dos componentes: Cordones epiteliales que a veces delimitan estructuras rosetoides y un estroma mixoide que recuerda al mesénquima embrionario (figura 18). Existe una variedad teratoide, en la que existen componentes heterólogo, cartílago y músculo estriado (32).
La mayoría son benignos o localmente agresivos pero cuando existen criterios histológicos de malignidad pueden metastatizar (33).
7. T. de células germinales * Teratoma: Es un tumor congénito poco frecuente (34) que se origina de una célula embrionaria pluripotencial capaz de desarrollar tejidos que derivan de las tres capas germinales: ectodermo (piel anejos dérmicos y derivados neuroectodérmicos), endodermo (epitelio gastrointestinal y respiratorio) y mesodermo (cartilago, hueso, músculo, tej. Adiposo y conectivo). Pueden ser quísticos o sólidos, generalmente son congénitos o aparecen en la infancia, suelen ser unilaterales, de crecimiento rápido y no se asocian a otras anomalías. Se comportan como tumores benignos aunque en un bajo porcentaje de teratomas existe un comportamiento maligno tipo tumor del seno endodérmico, que también puede aparecer «de novo» en la edad adulta sin que exista un teratoma acompañante, en este caso es de mal pronóstico por tener un comportamiento muy agresivo. Histológicamente se clasifican en maduros e inmaduros. Los inmaduros pueden presentar amplias áreas neuroectodérmicas, que plantean el diagnóstico diferencial con neuroblastomas y retinoblastomas, o mesenquima inmaduro tipo blastema metanéfrico o diferenciación rabdomioblástica. Los teratomas Maduros contiene estructuras ectodérmicas (epitelio escamoso, pelos y glándulas sebáceas, tej. nervioso, pexos coroides) y mesodérmicas (cartílago, hueso, etc) (figura 19).
8. T. de origen neuroepitelial El T. Neuroectodérmico melanocítico es poco frecuente, generalmente benigno, se presenta en el primer año de la vida y es también conocido bajo los nombres de tumor de anclaje retiniano o progonoma melanocítico. Es un tumor derivado de la cresta neural con marcada diferenciación hacia epitelio pigmentario y escasa maduración del componente neuroblástico (35). Ocurre preferentemente en el maxilar (36) y puede infiltrar secundariamente la órbita (37) aunque tambien se ha descrito su localización primaria en la órbita (39). Histológicamente está compuesto por túbulos o cordones de células pigmentadas, a veces con luces centrales, generalmente ocupadas por nidos de células pequeñas neuroblásticas. Esta población celular está incluida en un estroma conectivo denso (figuras 20 y 21).
El T. Neuroectodérmico periférico (PNET) comparte anormalidades géneticas y moleculares con el Ewing extraesquelético del que sólo se diferencia por presentar diferenciación neural. En la actualidad se consideran extremos polares de un mismo grupo tumoral. Histológicamente el PNET está compuesto por una proliferación de células redondeadas con escaso citoplasma y núcleos redondeados de cromatina dispersa finamente granular. En ocasiones diferencian estructuras rosetoides semejantes al neuroblastoma (figuras 22 y 23). Con inmunohistoquímica las células expresan positividad para marcadores neurales: neurofilamentos, sinaptofisina y cromogranina.
9. T. pigmentario En el epitelio conjuntival existe una población de melanocitos que pueden proliferar y dar lugar a nevus. Como sucede en la epidermis los nevus pueden ser junturales, compuestos o mixto. Los nevus conjuntivales presentan peculiaridades histológicas que los diferencian de los epidérmicos, se acompañan en su crecimiento de células epiteliales de reserva que pueden madurar como células escamosas o mucosas, cuando proliferan estas últimas delimitan estructuras pseudoglandulares (pseudoglándulas de Henle) que a veces configuran auténticos quistes (figura 24). La proliferación epitelial puede ser tan intensa que enmascare la proliferación névica. En algunos nevus conjuntivales existe una marcada infiltración linfocitaria alrededor de esta proliferación juntural.
Los melanomas conjuntivales en niños son muy raros (39) y de mal pronóstico.
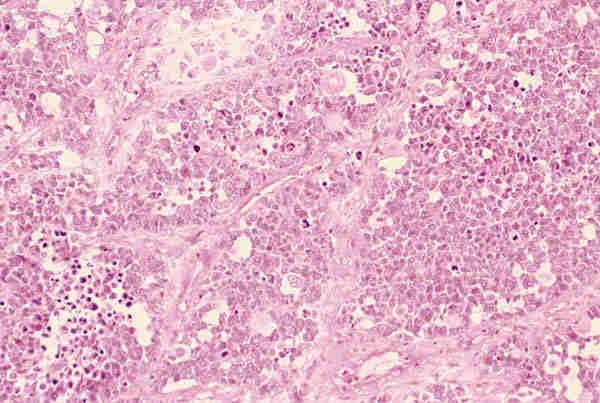
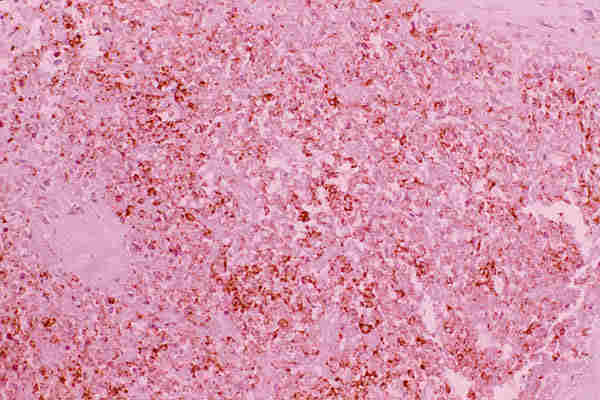
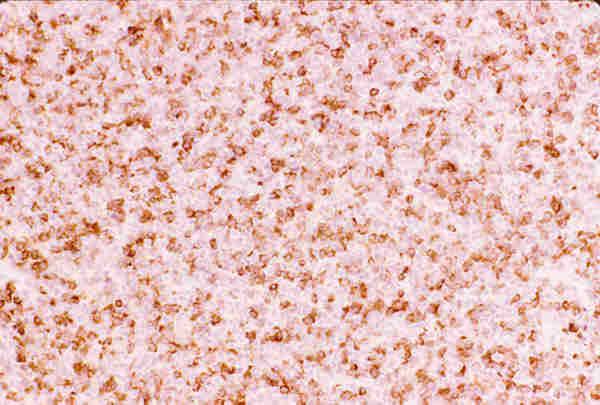
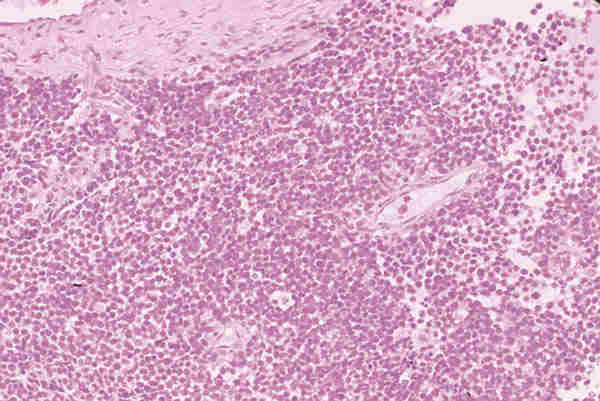
10. T. hematopoyéticos Los tumores de origen hematopoyético son raros y representan el 2,5 en algunas series antiguas (22,42). En series recientes se ha observado un notable aumento de procesos linfoides tumorales del adulto con nula incidencia en la edad pediátrica (29,43). Las leucemias pueden aparecer en los niños como tumores de partes blandas de situación orbitaria o palpebral y reciben el nombre de sarcoma mieloide, sarcoma granulocítico o cloroma, a veces preceden en semanas o meses a la infiltración de la médula osea (44,45). Histológicamente aparece una proliferación difusa de células de pequeño o mediano tamaño, con núcleos ovoides, a veces ligeramente lobulados y frecuentemente en mitosis, poseen citoplasmas acidófilos, a veces ligeramente granular (figuras 25 y 26) y expresan positividad con el antígeno leucocitario común (LCA) y la mieloperoxidasa.
11. T. óseos La F. osificante psamomatoide es un tumor óseo que hay que distinguir clínica e histológicamente de otras lesiones fibro-óseas, tales como displasia fibrosa, fibroma osificante y fibroma cementante. Clínicamente tiene un comportamiento agresivo y puede erosionar e invadir los tejidos circundantes con una marcada predilección por los huesos orbitarios (frontal y etmoidal). Suele aparecer en las primeras décadas y como tratamiento se aconseja una extirpación quirúrgica completa (46). Histológicamente se observa un estroma muy celular con estructuras esféricas de colágeno calcificado semejantes los cuerpos de psamoma (figura 27).
La displasia fibrosa del hueso puede ser monostótica o poliostótica. La lesión orbitaria suele ser monostótica y aparece en las tres primeras décadas de la vida, en su crecimiento puede producir un estrechamiento del canal del nervio óptico y alteraciones del drenaje del conducto lagrimal (47). Histológicamente se observan las consecuencias de un paro de la maduración del tejido óseo, con presencia de trabéculas de tejido óseo inmaduro a veces curvilíneas incluidas en un estroma fibroso muy vascularizado (figura 28).
El Sarcoma de Ewing primario de órbita es muy infrecuente (48) y siempre es obligatorio descartar un tumor metastásico (49). Histológicamente existe una proliferación de células pequeñas, basófilas, indiferenciadas que a veces muestran citoplasmas vacuolados PAS positivos que ocupan los espacios intertrabeculares (figura 29).
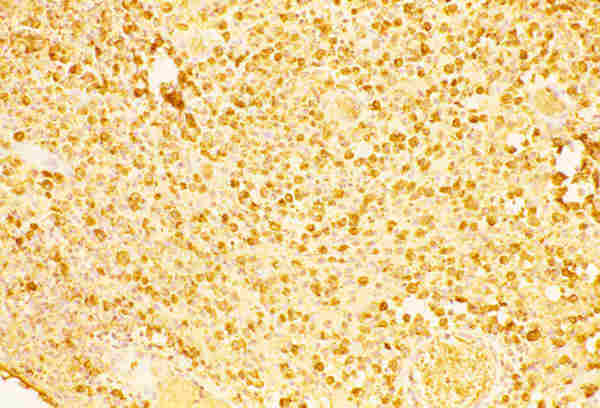
La histiocitosis X (granulomatosis de Langerhans) tiene, clínicamente tres formas de presentación: granuloma eosinófilo monostótico, enfermedad ósea multifocal (enf. Hand-shüller-Christian) y la enfermedad diseminada con afectación visceral (Enf. de Letterer-Siwe). Todos tienen en común la proliferación de histiocitos especializados en la presentación de antígeno (células de Langerhans). En la órbita puede manifestarse como una lesión solitaria o difusa. Histológicamente se observa una proliferación de histiocitos con núcleos hendidos o lobulados con citoplasma eosinófilo, a veces ligeramente granular, que expresan positividad con la tinción de inmunohistoquímica de la proteína S-100 y con el antígeno CD-1a (figuras 30 y 31). A veces se observa la presencia de células gigantes multinucleadas y de ocasionales eosinófilos.
El sarcoma osteogénico puede aparecer en pacientes que han sufrido radiaciones orbitarias (sobre todo en retinoblastomas).
TUMORES ORBITARIOS SECUNDARIOS En los niños las metástasis se localizan más frecuentemente en la órbita que en el globo ocular a diferencia de en los adultos donde la localización más frecuente es en la coroides El neuroblastoma es el tumor metastásico más frecuente en esta localización en la edad pediátrica (50). Se han descrito neuroblastomas orbitarios primarios en adultos pero no se ha publicado ninguno en niños (51). Cuando metastatizan en la órbita ya se conoce la localización primaria del tumor por lo que no se suelen biopsiar. Las leucemias, sobre todo la leucemia linfoblástica aguda (22), en fases avanzadas también metastatizan en la órbita. Los infiltrados orbitarios primarios (sarcomas granulocíticos) ya han sido comentados anteriormente.
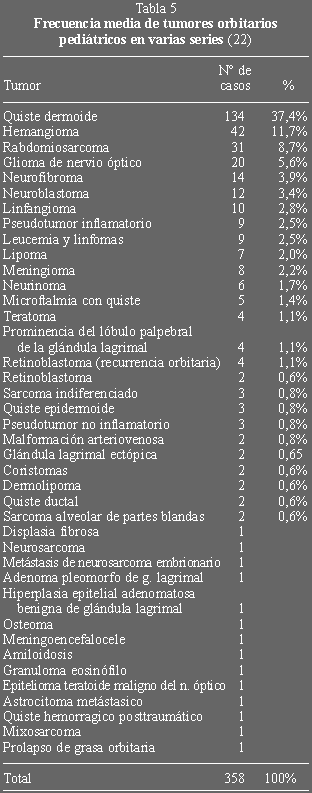
CASUÍSTICA Hemos revisado 159 estudios anatomopatológicos de lesiones tumorales y pseudotumorales orbitarias pediátricas recogidas durante el período comprendido entre 1967 y 2000 procedentes del servicio de oftamología de la ciudad sanitaria La Paz (tabla 4). En la tabla 5 se puede realizar un estudio comparativo basado en una recopilación de distintas series (22).
Hemos excluido las lesiones inflamatorias agudas y las crónicas inespecíficas incluyendo solo aquellas lesiones inflamatorias que desde el punto de vista clínico o anatomopatológico planteaban el diagnóstico de un proceso neoplásico.
Lesiones pseudotumorales inflamatorias Dentro de estos hemos encontrado 10 lesiones inflamatorias pseudotumorales (6,2% del total) que se distribuyen de la siguiente manera:
Lesiones pseudotumorales por anomalías del desarrollo Un total de 49 lesiones quedan incluidas en este apartado (30% del total) y su distribución es la siguiente:
Como en todas las series revisadas, los dermoides son lo más frecuentes (22,40) sus porcentajes oscilan entre el 30% y el 5% de todas las lesiones orbitarias. La mayoría de ellos corresponden a lesiones quísticas y uno de nuestros siete casos de dermoides sólidos formaban parte de un síndrome de Goldenhar. En cuatro casos las anomalías del desarrollo ocular dieron lugar a una microftalmia con quiste colobomatoso, unilateral en todos los casos y no existían otras malformaciones asociadas. Sólo uno de los casos no fue diagnosticado clínicamente y planteó el diagnóstico diferencial con una neoformación orbitaria. Como ejemplo de encefalocele hemos encontrado una heterotopía glial a nivel de la órbita en la que clínicamente no se demostró orificio de comunicación. Una marcada hiperplasia de la glándula lacrimal, planteó en uno de nuestros casos el diagnóstico diferencial con una neoplasia orbitaria. En cuatro casos la patología lacrimal consistía en una dilatación quística del conducto que podría corresponder a glándulas lacrimales ectópicas con fenómenos ectásicos.
Tumores orbitarios primarios Hemos identificado 94 lesiones primarias de la órbita. El rabdomiosarcoma es el proceso neoplásico más frecuente en la órbita, tanto en nuestro material (20 casos, 12,5%) como en las diferentes series publicadas (42). Según la nueva clasificación del Instituto Nacional del Cáncer (18) los hemos reclasificado en:
En 5 de nuestros casos hemos estudiado el tumor primario y la recidiva (1 R. alveolar y 4 R. embrionarios, uno de éstos mostraba áreas de agresividad citológica). Los rabdomiosarcomas convencionales mostraban en las recidivas amplias zonas de diferenciación celular. En otros dos casos no hemos tenido acceso al tumor primario por lo que sólo hemos estudiado las recidivas, una de ellas presentaba una marcada diferenciación celular, en el segundo caso existían amplias zonas de citología agresiva. El segundo grupo en frecuencia lo constituyen los tumores pigmentarios, 9 lesiones (11,9%) que se distribuyen en nevus conjuntivales (5 casos) y nevus compuestos los restantes 14 casos. En ninguno de nuestros casos existían nevus displásicos ni melanomas. Se han observado 16 casos de tumores con diferenciación vascular (10%), 10 de los cuales pertenecen a hemangiomas capilares en fase proliferativa, 4 casos presentaban una imagen de malformación linfática-venosa y 2 correspondían a hemangiomas cavernosos o a hemangiomas capilares en fase involutiva. En 8 casos de nuestro material hemos encontrado diferenciación neural, 7 correspondían a neurofibromas de patrón plexiforme y los enfermos presentaban un cuadro de neurofibromatosis. Hemos encontrado 1 caso de neurinoma pero no tenemos ningún neurofibrosarcoma orbitario en la edad pediátrica. A nivel del N. óptico hemos encontrado 4 tumores (2,5%), meduloepiteliomas 2 casos, glioma 1 caso y meningioma 1 caso. Diferenciación adiposa existió en 7 casos de nuestro material, en todos existía tejido adiposo maduro y diferenciado. Uno de los casos se acompañaba de prolapso de la grasa orbitaria. Encuadrables como tumores óseos hemos encontrado 10 casos (6,3%), 7 corresponden a granulomas eosinófilos monostóticos, y los restantes eran sarcomas osteogénicos postradiación de retinoblastoma, Ewing primario de órbita y fibrosis osificante psamomatoide. Los tumores hematopoyéticos primarios orbitarios han resultado poco frecuentes en nuestro material existiendo únicamente un sarcoma granulocítico. Como tumor germinal hemos encontrado un solo caso de teratoma diferenciado en el que no encontramos áreas de tumor del seno endodérmico. Como tumores de origen neuroepitelial hemos incluido 2 casos de tumor neuroectodérmico melanocítico y un caso de tumor neuroectodérmico periférico de localización orbitaria. Por último hemos incluido 4 lesiones epiteliales correspondientes a papilomas conjuntivales que también planteaban problemas con neoplasias orbitarias
Tumores orbitarios secundarios 1. Por extensión: En tres casos la órbita estaba infiltrada por un retinoblastoma, previamente a todos se les había extirpado el globo ocular y en dos de ellos, en la pieza quirúrgica, se comprobó infiltración del nervio óptico. Uno de nuestros casos presentaba la órbita infiltrada secundariamente por un rabdomiosarcoma alveolar originado en región malar (figura 32).
A nivel de la lámina cribosa de fosa nasal existía en uno de nuestros casos un estesioneuroblastoma que igualmente infiltraba la órbita.
2. Metastásicos: Sólo hemos encontrado un caso de metástasis de neuroblastoma que planteó diagnóstico diferencial con una neoplasia orbitaria primaria.
BibliografÍa
|