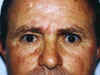
 Fig. 1
Fig. 1Dres. García E1, Casado R2, Machín S3, Pérez-Torregrosa VT4
Hospital de Hellín. Albacete.
(1) MIR de Medicina Familiar y Comunitaria.
(2) FEA. Servicio de Oftalmología.
(3) Médico de Familia.
(4) Jefe de Sección. Servicio de Oftalmología.
Introducción
Las escleras azules son un hallazgo poco frecuente en la práctica oftalmológica habitual. Su presencia puede asociarse o no, a fragilidad ósea con fracturas, anomalías de la dentición y sordera, formando parte de un síndrome de osteogénesis imperfecta.
El fundamento histológico de esta coloración es un adelgazamiento de la esclera que permite la transparencia del pigmento uveal dando así un color azul a las escleróticas.
Presentamos el caso de un paciente con escleras azules como hallazgo inicial que permitió el diagnóstico de una osteogénesis imperfecta familiar.
Caso clínico
Un varón de 43 años acude a una exploración rutinaria a la consulta de Oftalmología detectándose unas escleras azules bilaterales (fig. 1) sin otra patología ocular.
Fig. 1
En la historia clínica del paciente destacan hipoacusia del oído izquierdo, fractura de codo a los 10 años y fractura distal de radio acortada e impactada a los 35 años.
Entre las exploraciones complementarias realizadas al paciente se encuentran varias audiometrías (sin diferencias significativas entre las mismas) y una densitometría ósea con riesgo medio de fractura-luxación. El resto de la exploración fue normal.
Al estudiar a los familiares, para determinar la sintomatología acompañante de las escleras azules y el patrón de herencia familiar, se encuentra que la abuela presentaba sordera y escleras azules; tanto el paciente como su padre presentaban sordera, fragilidad ósea y escleras azules (esquema 1).
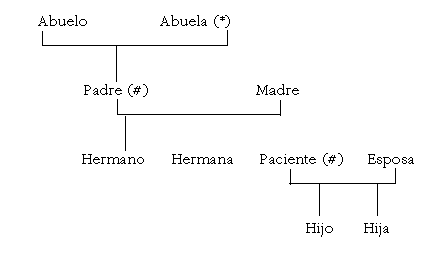
Esquema 1. Genograma.
(*): sordera, escleras azules.
(#): sordera, fragilidad ósea, escleras.
Discusión
El término de esclera azul se debe al adelgazamiento de las fibras que componen la esclerótica lo que permite la transparencia de los vasos coroideos a su través. La presencia aislada de escleras azules se debe a un trastorno estructural del tejido conectivo (1) o a un defecto de la pigmentación ocular. En ocasiones (sobre todo en niños) se ha relacionado con anemia (2).
Nuestro paciente tiene las escleras azules asociadas a una hipoacusia congénita del oído izquierdo, que no ha empeorado a lo largo del tiempo, y a dos fracturas óseas. La exploración con lámpara de hendidura no reveló la existencia de alteraciones macroscópicas de la córnea (queratoglobo, queratocono) que suelen aparecer asociadas a escleras azules de herencia autosómica recesiva (3,4). Al mover la conjuntiva no hay cambios de coloración con lo que se descarta la presencia de áreas de pigmentación conjuntival (5) (fig. 2).
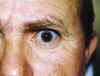 Fig. 2
Fig. 2
En la osteogénesis imperfecta hay una alteración del tejido conectivo, que se manifiesta con fragilidad ósea, escleras azules y sordera. En la misma se ha observado con microscopia óptica una disminución del diámetro de las fibras de colágeno tipo I y la desaparición de las estrías transversales (6), produciéndose la desorganización del tejido y la transparencia de la coroides.
La osteogénesis imperfecta se puede clasificar en diferentes tipos (I-IV de Sillence) según la herencia:
Los dos últimos se caracterizan por un aumento progresivo de la gravedad del síndrome, pero sin escleras azules en la edad adulta (7,8).
Nuestro paciente pertenece al tipo I porque, junto a un patrón de herencia dominante (los portadores están enfermos, la mitad de la descendencia estará enferma y la enfermedad aparecerá en todas las generaciones), presenta una clínica de escleras azules y sordera congénita. Sus hijos están asintomáticos, lo cual podría deberse a una mutación en el tipo de herencia (9).
El diagnóstico diferencial de la osteogénesis imperfecta se realiza con una serie de patologías (tabla I). Siempre habrá que descartar algunos signos característicos como el síndrome de ligamentos laxos aislado, el síndrome de «brittle cornea» (córnea «reluciente» y pelo rojo) (10), el síndrome de Cornelia de Lange (retraso mental, retraso del crecimiento y anomalías esqueléticas sin escleras azules) (11), el síndrome de Marfan congénito (megalocórnea, iris alterado y mal desarrollo de los cuerpos ciliares) (12).

No existe tratamiento curativo ni de las escleras azules, puesto que son una manifestación de la anomalía histológica, ni de la fragilidad ósea. Sin embargo, con medidas preventivas y paliativas (consejo genético, educación, ejercicios de rehabilitación, ) la mayoría de los pacientes consiguen una calidad de vida aceptable.
De lo expuesto destacamos que ante todo paciente con escleras azules es importante descartar otra patología subyacente. Además, al ser una enfermedad hereditaria es imprescindible el estudio de la familia para poder dar consejo genético y calcular el riesgo de los descendientes de padecer la enfermedad.
Agradecimientos
Deseamos agradecer al Dr. Francisco García-Pourriños (Servicio de Otorrinolaringología) y al Dr. Isidro Tormos (Servicio de Oftalmología) su colaboración en el estudio de este caso.
Resumen
Tras el hallazgo casual de un paciente con escleras azules en una visita rutinaria al Servicio de Oftalmología, se efectúa un estudio familiar que abarca cuatro generaciones.
Se realiza un análisis retrospectivo y un estudio prospectivo. Aparecen asociados sordera y fragilidad ósea en varios de los miembros y se llega al diagnóstico de osteogénesis imperfecta. Ante el hallazgo de escleras azules es imprescindible un estudio ocular y sistémico para descartar patología asociada.
Palabras clave
Escleras azules, sordera, fractura ósea, osteogénesis imperfecta.
Summary
After the fortuitous finding of blue esclerotics in a patient during an ordinary visit to the Ophthalmologist, a family study was done along four generations.
A retrospective analysis and prospective study were done. There was an association between deafness and bone fractures in some of the family memberships, so it was diagnosed as Imperfect Osteogenesis.
When blue esclerotics are found in a patient, an ophthalmologic and systemic study is needed in order to discard associated pathology.
Key words
Blue esclerotics, deafness, bone fractures.
Bibliografía