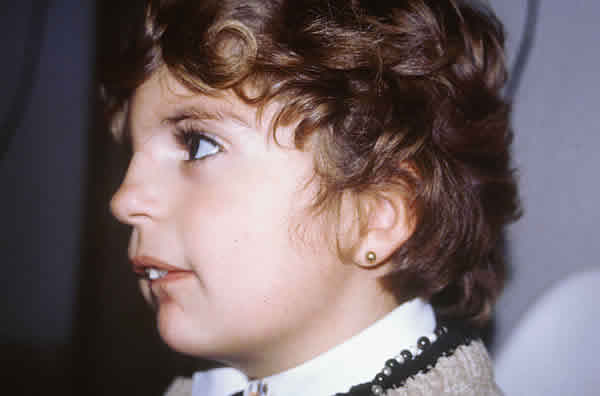
Fig. 1. Vista lateral: micrognatismo que le da el aspecto de cara de pájaro.
Dres. Asensio Sánchez VM1, De Paz M2, Lamarca Pinto E3, Rodríguez Concellón L2
Servicio de Oftalmología. Hospital General
INSALUD. Medina del Campo. Valladolid.
(1) Doctor en Medicina y Cirugía.
(2) MIR 3.er año.
(3) MIR 2.º año.
Introducción
El síndrome de Pierre Robin es una anomalía congénita del arco branquial en la que se asocia micrognatismo, glosoptosis y hendidura del paladar, por lo que estos niños desarrollan desde el nacimiento problemas respiratorios y digestivos severos que se pueden asociar a mortalidad elevada (1,2).
Al ser una anomalía del primer arco branquial, se asocian múltiples problemas oftalmológicos que pueden pasar desapercibidos al clínico (pediatras, cirujanos pediátricos y oftalmólogos) ante la gravedad del cuadro sistémico, pero que pueden comprometer el posterior desarrollo del niño (1,3).
Se presenta un caso de síndrome de Pierre Robin tratado en nuestro servicio desde el nacimiento, que en la actualidad tiene 4 años, y que presentó unas características oftalmológicas diferenciales con los casos descritos en la literatura.
Caso clínico
Niña sin antecedentes familiares de interés, salvo que la madre fue fumadora durante los primeros meses del embarazo; nace en un parto eutócico a las 37 semanas de gestación y con un peso de 2,650 gramos. En la exploración neonatal se aprecia dificultad respiratoria severa con cianosis distal, por lo que ingresa en la UVI pediátrica por su situación crítica, donde se inferior, glosoptosis con lengua de tamaño normal y el paladar hendido, lo que dificultaba la respiración y la alimentación. A los tres meses se realiza un estudio oftalmológico donde se aprecia telecanto con esotropía bilateral de 20° y megalocórneas que medidas en quirófano tienen un diámetro horizontal de 13 mm y tensiones de 12 mmHg en ambos ojos; en la misma anestesia se inyectó 7 UI de toxina botulínica (Botox) en cada recto medio. En el estudio biomicroscópico destacan las escleras azules, con un cristalino normal en forma y posición, así como el estudio de fondo. La refracción de la paciente es de +5 D en el ojo derecho y +5,5 D en el ojo izquierdo.
Se alimentó a la niña con una sonda gástrica implantada hasta los 16 meses y se le operó 3 veces del paladar para cerrarlo, llevar la lengua a su posición original y favorecer la dentición. Actualmente tiene 4 años edad, y aunque presenta estigmas característicos de la enfermedad (figs. 1 y 2), desarrolla una vida escolar y social normal.
Fig. 1. Vista lateral: micrognatismo que le da el aspecto de cara de pájaro.
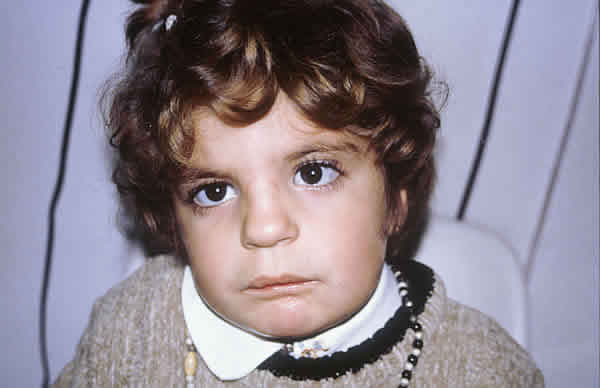
Fig. 2. Vista frontal: telecanto, escleras azules.
Nótese el reflejo de la luz sobre ambas córneas y la marca de las gafas de
hipermetropía fuerte.
Discusión
Fue Pierre Robin el que describió por primera vez este síndrome, aunque posteriormente se consideró que la micrognatia, la glosoptosis y la hendidura palatina eran todos secuencia de una hipoplasia inicial en el desarrollo mandibular, por lo que muchos autores han sugerido el cambio del término síndrome por el de secuencia de Pierre Robin o complejo de Robin influido por la variabilidad fenotípica (1,4).
El síndrome de Pierre Robin es una alteración en el desarrollo embrionario del primer arco braquial en las primeras nueve semanas de la gestación, que origina una hipoplasia en la mandíbula con inserción posterior de la lengua, que es de tamaño normal, impidiendo el cierre del paladar distal, con lo que no se produce el desarrollo correcto del macizo cráneo-encefálico con dificultad respiratoria y en la deglución (1); por todo esto la secuencia de Pierre Robin se clasifica dentro de las anomalías esqueléticas al ser esencialmente una patología de la cavidad oral posterior (4,5).
Este síndrome generalmente ocurre como caso aislado sin patología asociada, aunque tampoco es raro que se asocie a otros problemas sistémicos (neurológicos, cardíacos) y más frecuentemente oftalmológicos (1,5,6), por lo que el oftalmólogo debe tener conocimiento de este proceso.
Las anomalías oculares descritas son numerosas, destaca por su frecuencia la esotropía y el glaucoma congénito que requieren ambas un tratamiento urgente, pues estos niños cuando sobrepasan los 4 años de edad van a desarrollar una vida completamente normal con un correcto desarrollo socio-escolar como es el caso que presentamos (3), de aquí la importancia de reconocer esta patología en los primeros momentos; el problema se puede agravar cuando se presenta una megalocórnea, lo que obliga a estudios bajo anestesia general con el riesgo que supone en estos recién nacidos como ocurrió en esta paciente (5). Es importante descartar la asociación descrita entre Pierre-Robin y aniridia (6) por el pronóstico vital que puede implicar. Nuestro caso tenía las escleras azules, dato que no aparece recogido en la bibliografía asociado a este síndrome y que desconocemos su significación. Otra característica diferencial de nuestra paciente con los presentados en la literatura es la presencia de telecanto, proceso compatible con una alteración del primer arco branquial (1,4). Estos niños pueden desarrollar cataratas congénitas y luxación de cristalino hasta en el 33% de los casos (7), por lo que se requiere una exploración oftalmológica precoz.
Otro dato diferencial con respecto a la bibliografía es que esta niña presentaba una hipermetropía alta, pero siempre se debe estudiar el polo posterior ante la posibilidad de grandes miopías (que es lo más frecuente), degeneraciones vítreo-retinianas y desprendimientos de retina (1).
En conclusión, presentamos un caso de complejo de Pierre Robin con características diferenciales con respecto a los descritos en la literatura: escleras azules, hipermetropía, telecanto y megalocórnea que en un principio puede confundirse con otros síndromes.
Resumen
La descripción típica del síndrome de Pierre Robin incluye micrognatismo, glosoptosis, obstrucción de las vías aéreas y en la mayoría de los casos fisura palatina. Actualmente la consideración de este síndrome ha cambiado agrupando retrognatia, fisura palatina y distress respiratorio.
La clínica oftalmológica fundamental en el síndrome de Pierre Robin incluye el glaucoma congénito y miopía magna congénita responsable de desprendimiento de retina. Presentamos un caso con un seguimiento de cuatro años que como datos diferenciales con lo descrito en la literatura presenta megalocórnea, escleras azules, telecanto e hipermetropía fuerte.
Palabras clave
Pierre Robin, megalocórnea, esclera azul, telecanto, hipermetropía.
Summary
The classical description of the Pierre Robin syndrome includes micrognathia, glossoptosis, airway obstruction, and usual presence of a cleft palate. The Pierre Robin syndrome is currently defined as the combination of retrognathia, cleft palate, and respiratory distress.
The main ocular manifestations found in the Pierre Robin syndrome are congenital glaucoma and severe congenital myopia responsable for retinal detachment.
We present a case examine during four with the next different characteristic megalocornea, blue sclera, telecanthus and high hypermetropia.
Key words
Pierre Robin, megalocornea, blue sclera, hypermetropia.
Bibliografía
Feingold M, Gellis SS. Ocular abnormalities associated with first and second arch syndromes. Surv Ophthalmol 1969 Jul; 14(1): 30-42.
Cruz MJ, Kerschner JE, Beste DJ, Conley SF. Pierre Robin sequences: secondary respiratory difficulties and intrinsic feeding abnormalities. Laryngoscope 1999 Oct; 109(10): 1632-1636.
Kapp-Simon KA. Krueckeberg Mental development in infants with cleft lip and/or palate. SCleft Palate Craniofac J 2000 Jan; 37(1): 65-70.
Cohen MM Jr. Robin sequences and complexes: causal heterogeneity and phatogenetic/phenotypic variability. Am J Med Genet 1999 Jun 4; 84(4): 311-315.
Barker I. Anaesthesia for Pierre-Robin syndrome. Hosp Med 2000 Jan; 61(1): 72.
Camassei FD, Jenkner A, Bertini E, Bosman C, Donfrancesco A, Boldrini R. Pierre Robin syndrome and Wilms tumor: an unusual association. Med Pediatr Oncol 2000 Jul; 35(1): 83-84.
Rogers NK, Strachan IM. Pierre Robin anomalad, maculopathy, and autolytic cataract. J Pediatr Ophthalmol Strabismus 1994 Nov-Dec; 32(6): 391-392.