Fig. 1. Árbol genealógico en la paciente. En rojo los familiares afectos de patología retiniana. La flecha señala a la paciente.
CASO CLÍNICO
MONTERO MORENO JA1, RUIZ MORENO JM2, DE LA VEGA GALIANA C1
(1) Doctor en
Medicina y Cirugía. Instituto Oftalmológico de Alicante, VISSUM. Unidad de
Retina Vítreo.
(2) Doctor en Medicina y Cirugía. Departamento de Oftalmología. Universidad
Miguel Hernández de Alicante. Profesor Titular.
RESUMEN
Introducción: La retinosquisis familiar es una enfermedad de herencia habitualmente ligada al cromosoma X. Uno de sus aspectos más característicos es la retinosquisis macular.
Caso clínico: Una mujer de 40 años con antecedentes familiares de patología retiniana no bien filiada, consultó por disminución de agudeza visual. A la exploración se pudo comprobar la presencia de una cavidad en área macular, sin patología retiniana periférica. Se realizó un estudio angiográfico y electrofisiológico que confirmaron el diagnóstico. Igualmente se realizó un estudio mediante tomografía óptica de coherencia (OCT 3). Se describen los hallazgos.
Discusión: Se discute el diagnóstico diferencial con otros cuadros, así como la contribución de la OCT 3 en el estudio de este paciente.
Palabras claves: Retinosquisis macular, OCT.
SUMMARY
Introduction: Juvenile familiar retinoschisis is commonly associated to X linked inheritance. Macular retinoschisis is considered to be a hallmark of this condition.
Case report: A 40 year old female patient, with familiar history of an inherited retinal disorder, complained of progressive visual loss. Biomicroscopy revealed the presence of macular retinoschisis without significant changes in retinal periphery. An angiographic and electrophysiological study were performed which confirmed the diagnosis. An optical coherence tomography (OCT 3) study was performed.
Discussion: Differential diagnosis is discussed, as well as the role of OCT 3 in the evaluation of this patient.
Key words: Macular retinoschisis, OCT.
INTRODUCCIÓN
La retinosquisis macular (RM) se considera un factor característico de la retinosquisis juvenil familiar ligada al cromosoma X, aunque actualmente se sabe que puede asociarse a múltiples enfermedades tanto hereditarias como adquiridas (1). Salvo raras excepciones, la RM se presenta solamente en el sexo masculino. La mácula se encuentra afectada en todos los casos, y en muchos pacientes se puede comprobar además la presencia de múltiples defectos en la retina periférica y en el epitelio pigmentario de la retina (EPR). Biomicroscópicamente la RM muestra un cuadro característico con pequeños quistes en la superficie de la retina conformando un patrón estrellado y con estrías radiales centradas en la fóvea. Con el tiempo las cavidades quísticas pueden coalescer y originar una gran cavidad quística. Algunos pacientes pueden mostrar un estrechamiento progresivo de los vasos retinianos, y cambios periféricos similares a las distrofias retinianas pigmentarias (2). Pese a que el patrón ligado a X es el más frecuente, se han descrito también otros patrones hereditarios (3).
CASO CLÍNICO
Una mujer de 40 años de edad con antecedentes familiares masculinos y femeninos de patología retiniana no bien filiada, acudió a la consulta describiendo una disminución progresiva de agudeza visual a lo largo del último año. El árbol genealógico (fig. 1) mostró un patrón de herencia autonómico dominante. La agudeza visual corregida era de 0,2 en ambos ojos. No existía dificultad en la adaptación a la oscuridad. Ambos ojos mostraban un patrón macular estrellado característico de la RM, sin presencia de otras alteraciones quísticas ni defectos pigmentarios en polo posterior ni en periferia retiniana.
![]()
Fig. 1. Árbol genealógico en la paciente. En rojo los familiares afectos de
patología retiniana. La flecha señala a la paciente.
La angiografía fluoresceínica (AF) reveló una hiperfluorescencia macular precoz que se mantuvo a lo largo de los tiempos angiográficos, sin extravasación de fluoresceína, correspondiéndose con un efecto ventana. La campimetría computarizada mostraba una discreta reducción concéntrica del campo en ambos ojos.
Se realizó una tomografía óptica de coherencia (OCT 3), que demostró la presencia de cavidades quísticas radiales coalescentes en área macular que originaban una separación de las capas internas de la retina en ambos ojos limitada al área foveal. No se apreciaron tracciones vítreas. Igualmente se pudo constatar un engrosamiento foveal en el ojo izquierdo a expensas de las cavidades quísticas, mientras que en el ojo derecho el espesor foveal se hallaba reducido (figs. 2 y 3).
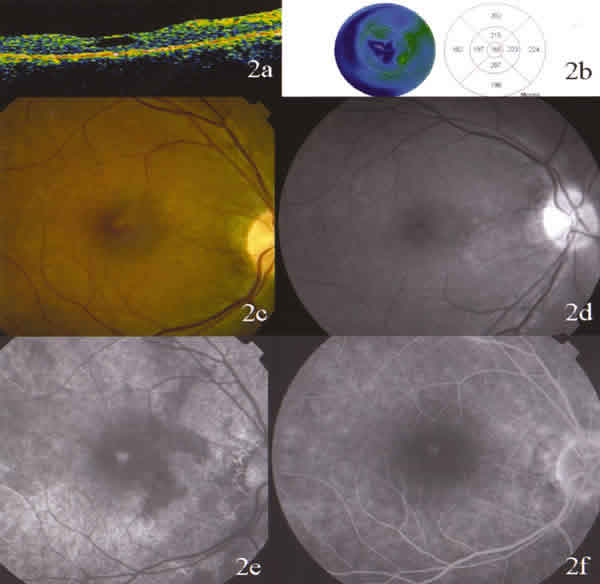
Fig. 2. Ojo derecho. 2a: Imagen de OCT de retinosquisis macular. Se puede
apreciar la presencia de una cavidad quística que se corresponde con la fóvea.
2b: mapa topográfico que muestra una depresión en el centro de la mácula. 2c: la
retinografía revela cambios pigmentarios y pliegues maculares radiales en "rueda
de carro". 2d: fotografía aneritra que demuestra los mismos hallazgos. 2e: los
tiempos angiográficos precoces muestran la hiperfluorescencia del área quística.
2f: los tiempos tardíos no revelan cambios en la hiperfluorescencia ni
extravasación de la fluoresceína.

Fig. 3. Ojo izquierdo. 3a: imagen de OCT de retinosquisis macular. Cavidad
quística en la fóvea. 3b: topografía OCT elevación del centro de la mácula. 3c y
3d: retinografía y aneritra con pliegues radiales centromaculares. 3e y 3f:
tiempos precoces y tardíos, similares al ojo derecho.
El electrorretinograma con flash mostraba una respuesta bilateral bien definida para la luz azul y blanca, y una respuesta más atenuada, aunque detectable, para la luz roja. La onda a era simétrica en ambos ojos y la onda b estaba atenuada especialmente en el ojo derecho (onda b ojo derecho 38,7 microvoltios, ojo izquierdo 88,0 microvoltios).
DISCUSIÓN
Recientemente se han publicado los hallazgos mediante OCT en ojos con RM y con retinosis pigmentaria (RP) (4-7). En el caso que se describe, se ha podido comprobar la presencia de una separación de las capas de la retina y la formación de cavidades quísticas intrarretinianas que no mostraban acúmulo de fluoresceína, patrón éste que se corresponde con el de la RM. No se pudo objetivar la presencia de tracciones vítreas mediante biomicroscopia ni mediante OCT.
Nuestra paciente mostraba una forma atípica de RM confirmada mediante OCT y AF. No presentaba antecedentes familiares claros de retinosquisis ni de consanguinidad. En este caso se pudo descartar el diagnóstico de una retinosquisis familiar ligada a X por su sexo y por carecer de antecedentes familiares claros, si bien no se pueden excluir otras formas de retinosquisis familiar con patrones hereditarios diferentes. No se identificaron signos de atenuación vascular ni de palidez papilar sugerentes de retinosis pigmentaria (RP), y de igual modo las alteraciones electrofisiológicas eran muy sugerentes de RM y no de RP.
Se ha descrito la presencia de RM en algunas familias con RP, especialmente en forma de edema macular, y con menos frecuencia con el patrón de una retinosquisis juvenil. En este caso también se pudo excluir la presencia de de un edema macular por la falta de extravasación durante la AF (6,7). La hiperfluorescencia precoz y tardía, en forma de efecto ventana, se atribuyó al defecto del tejido retiniano en la fóvea causado por las cavidades quísticas.
Atendiendo a la historia familiar de la paciente, y a su estado anatómico y funcional, consideramos que se puede tratar de una forma atípica de RM, de herencia autonómica dominante y sin retinosquisis periférica. La OCT 3 resultó de utilidad a la hora de identificar y caracterizar la enfermedad.
BIBLIOGRAFÍA
Madjarov B, Hilton GF, Lee SS. A new classification of the retinoschisis. Retina 1995; 15: 282-285.
Gass JDM. Stereoscopic Atlas of Macular Diseases: Diagnosis and Treatment. St Louis, Mo: Mosby, 4a ed. CD ROM.
Autosomal dominant inheritance of retinoschisis. Am J Ophthalmol 1982; 94: 338-343.
Azzolini C, Piero L, Codenotti M, Brancato R. OCT images and surgery of juvenile macular retinoschisis. Eur J Ophthalmol 1997; 7: 196-200.
Menchini U, Brancato R, Virgili G, Pierro L. Unilateral macular retinoschisis with stellate foveal appearance in two females with myopia. Ophthalmic Surg Lasers 2000; 31: 229-232.
ffytche TJ. Cystoid maculopathy in retinitis pigmentosa, Trans Ophthalmol Soc UK 1972; 92: 265-283.
Fishman, GA, Fishman, M, and Maggiano, J: Macular lesions associated with retinitis pigmentosa, Arch Ophthalmol 1977; 95: 798-803.