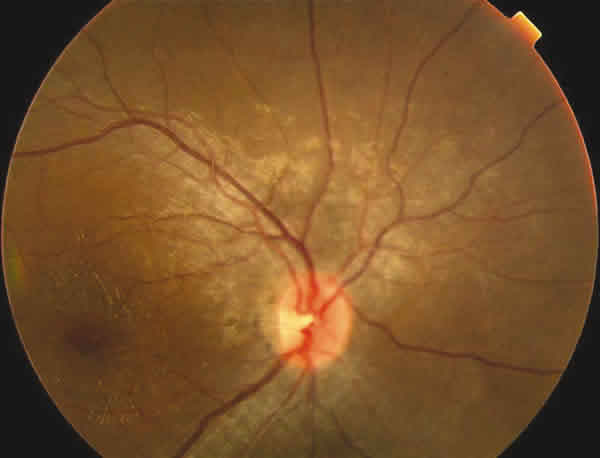
Fig. 1. Retinografía del ojo derecho.
SEMINARIO DE CASOS CLÍNICOS
Retinitis pigmentosa sine pigmenti
SUÁREZ BARAZA J1, GUTIÉRREZ DÍAZ A2
Hospital Universitario 12 de Octubre.
Madrid.
(1) Licenciado en Medicina.
(2) Doctor en Medicina.
RESUMEN
Caso clínico: Se presenta la secuencia diagnóstica de una paciente de 16 años con retinosis pigmentaria sin pigmento y nictalopia.
Discusión: En pacientes con signos mínimos de retinosis pigmentaria el diagnóstico puede ser orientado erróneamente, confundiéndose con procesos inflamatorios, neurológicos o glaucomatosos.
Palabras clave: Retinosis pigmentaria, retinosis pigmentaria sin pigmento, síndromes mascarada de uveitis.
SUMMARY
Case report: We report a diagnostic sequence of a sixteen years female with night blindness and minimal evidence of pigment migration into the retina
Discussion: The diagnostic in patients with few signs of retininitis pigmentosa could be erroneous because is easily confused with glaucoma and neurologic or inflamatory diseases.
Key Words: Retinitis pigmentosa, retinitis pigmentosa sine pigmenti, uveitis masquerade syndromes.
INTRODUCCIÓN
La retinosis pigmentaria sin pigmento pertenece junto a otras formas incompletas de retinosis pigmentaria (RP) al grupo de las distrofias retinianas pigmentarias atípicas. Para muchos autores representa el inicio de la típica RP (1).
CASO CLÍNICO
Paciente ecuatoriana de 16 años que consultó por déficit de agudeza visual (AV) en ambos ojos (AO).
Entre sus antecedentes personales destacaba un trasplante renal en su país por una insuficiencia renal crónica idiopática. Se encontraba en tratamiento con corticoides y micofenolato mofetilo por rechazo del injerto.
La AV corregida (1 a 20º en OD y 1,50 a 120º en OI) era 0,5 y 0,4 respectivamente.
La biomicroscopía anterior y la PIO eran normales.
En la oftalmoscopía llamaba la atención un reflejo grisáceo con un aspecto moteado del epitelio pigmentario retiniano (EPR). Tenía una ligera palidez papilar temporal en OI (figs. 1-3).
Fig. 1. Retinografía del ojo derecho.
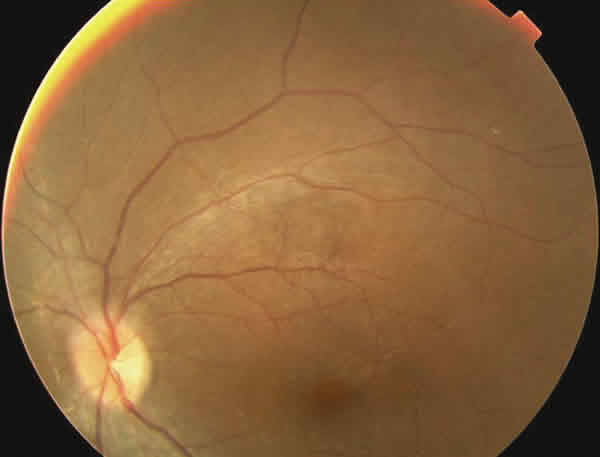
Fig. 2. Retinografía del ojo izquierdo.
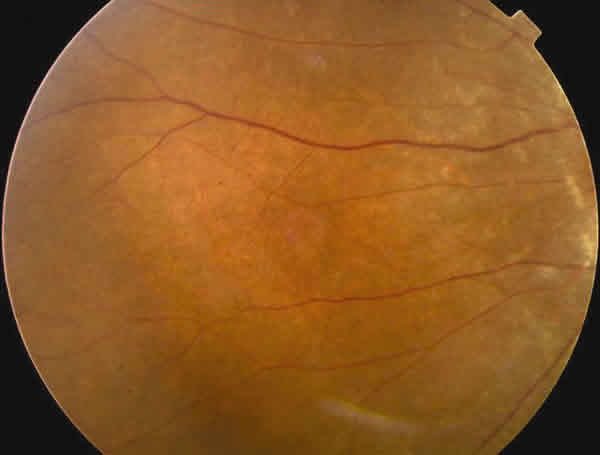
Fig. 3. Retinografía del ecuadro del ojo izquierdo.
Presentaba un tyndall vítreo anterior pigmentario de 1-2 + y llamaba la atención la hialinización de algunos vasos periféricos que simulaban envainamientos vasculares en AO (fig. 4). Mediante indentación escleral se descartó pars planitis.
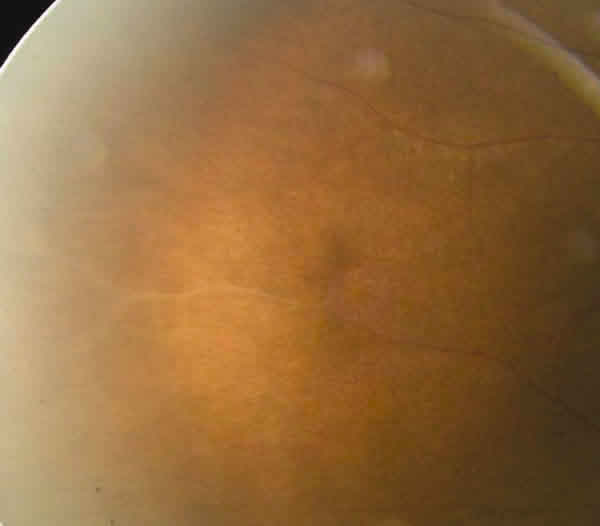
Fig. 4. Imagen de la periferia del ojo derecho que muestra la hialinización
vascular.
No se solicitó angiografía fluoresceínica (AFG) debido a la mala función renal de la paciente.
En la perimetría computarizada 30/2 (Humphrey instruments, modelo 740, Carl Zeiss Inc) se apreciaba una reducción concéntrica del campo visual con un islote central de 10º respetado que no ha empeorado en tres años de seguimiento (figs. 5 y 6). No obstante si ha desarrollado defectos de sensibilidad maculares en los campos visuales de umbral macular (fig. 7).

Fig. 5. Campimetría del ojo derecho. Reducción concéntrica que respeta 10º
centrales.
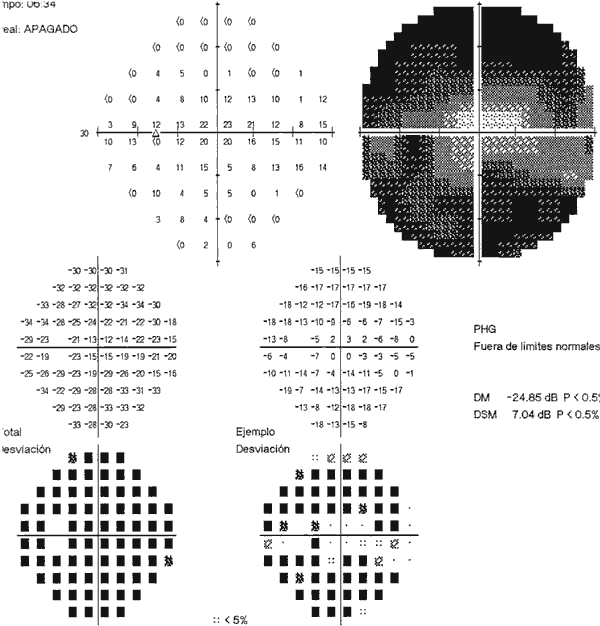
Fig. 6. Campimetría del ojo izquierdo. Reducción concéntrica.

Fig. 7. Perimetría de umbral macular de OD con algunos defectos de sensibilidad.
Al ser preguntada sobre su visión nocturna la paciente refería nictalopia desde la infancia que según ella no había empeorado.
En el estudio electrofisiológico el electrorretinograma (según normativa ISCEV) escotópico, fotópico y mesópico estaba abolido en AO.
No presentaba alteraciones cromáticas en el test de colores 28 Hue de Roth según Farnsworth-Munsell.
En el estudio familiar (padres y hermana) no se detectaron enfermedades retinianas degenerativas.
Se catalogó como una retinosis pigmentaria (RP) sin pigmento y en tres años de seguimiento permanece estable, no habiendo aparecido los signos oftalmoscópicos típicos de la RP (fig. 8).
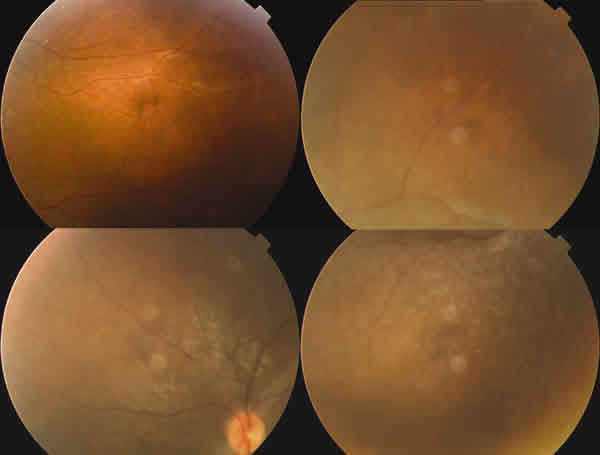
Fig. 8. Retinografías de AO tres años después del diagnóstico en las cuales
todavía no han aparecido los signos típicos de la retinosis pigmentaria. Se
aprecia el aspecto moteado del EPR a nivel ecuatorial.
DISCUSIÓN
La retinosis pigmentaria (RP) es una distrofia retiniana con signos y síntomas muy definidos entre los que se encuentran nictalopia progresiva, contracción de los campos visuales periféricos, estrechamiento vascular y el típico patrón de pigmento en espículas óseas.
Los primeros cambios oftalmoscópicos son la aparición de un reflejo grisáceo a nivel del EPR así como la despigmentación progresiva del mismo (1).
Existen diversas formas atípicas entre las que se encuentra la variedad sin pigmento, caracterizada por una migración pigmentaria mínima no detectable oftalmoscopicamente.
Los pacientes con la variedad sin pigmento suelen tener un déficit de visión nocturna menos severo así como menor afectación campimétrica y electrorretinográfica, lo cual no se corresponde con nuestra paciente (1-3).
Para otros autores la ausencia de migración pigmentaria no representa un subtipo específico de RP sino un estadio inicial de la enfermedad, pudiendo existir incluso décadas antes de aparecer las típicas características oftalmoscópicas, coexistiendo con escotomas relativos en la campimetría (4).
La angiografía es útil para detectar las zonas de despigmentación del EPR no visibles oftalmoscopicamente, pero la función renal de la paciente no nos permitió realizarla. Una AFG nos hubiera mostrado probablemente un edema macular incipiente como causa del déficit visual. La mayoría de los pacientes con edema macular cistoide tienen células en vítreo (1).
Investigaciones recientes revelan que la mitad de los pacientes en el momento del diagnóstico tienen alteraciones maculares, siendo mayor la incidencia en jóvenes con signos de mínima migración pigmentaria retiniana (1).
Dentro de las enfermedades renales que se han descrito asociadas a RP en algunas familias se encuentra el síndrome de Alport pero la falta de los típicos flecos perimaculares, las alteraciones electrofisiológicas y un nivel auditivo normal descartan tal síndrome (5).
Tambien se descartó la retinosis punctata albescens, una variedad de RP atípica que cursa con nictalopia y un clásico punteado amarillento en el EPR ecuatorial (1).
En pacientes con signos mínimos de RP el diagnóstico puede ser orientado de forma errónea, confundiéndose con procesos inflamatorios del segmento posterior (síndrome mascarada) ya que pueden presentar signos comunes a formas de uveítis intermedia como celularidad vítrea, catarata subcapsular posterior, periflebitis y edema macular cistoide (6,7).
En esta paciente hallamos celularidad vítrea pigmentaria procedente de la despigmentación del EPR y vasos retinianos periféricos hialinizados que simulaban envainamientos vasculares. Estos envainamientos también han sido descritos en retinosis pigmentaria, siendo el origen probablemente autoinmune ya que se han detectado respuestas autoinmunes en muchas enfermedades oculares degenerativas pero el papel en la patogénesis de la misma es desconocido (8).
En estudios del vítreo de pacientes con RP se han encontrado células del epitelio pigmentario, melanocitos uveales, astrocitos, linfocitos, y macrófagos. Podría ser el resultado de una reacción inmunológica secundaria frente a los antígenos retinianos vertidos en vítreo al degenerarse la retina (1,9).
Muchos pacientes afectos de RP no advierten los síntomas hasta que la visión central se afecta, lo cual unido a la ausencia de migración pigmentaria y la presencia de signos propios de las uveítis hace que el diagnóstico se pueda orientar de forma errónea. Pueden ser vistos inicialmente con bajas AV secundarias a edema macular cistoide o defectos campimétricos que simulan escotomas arciformes, confundiéndose con enfermedades neurológicas o glaucoma.
Algunos signos como la palidez papilar, el reflejo grisáceo del EPR y el estrechamiento de arteriolas deberían alertarnos para solicitar estudios electrofisiológicos.
BIBLIOGRAFÍA
Gass JDM. Heredodystrophic disorders affecting the pigment epithelium and retina. In:Gass JDM.Stereoscopic atlas of macular diseases:diagnosis and treatment on CD ROM. St Louis:Mosby; 1999; Vol. 2, chap 5.
Pearlman JT, Flood TP, Seiff SR. Retinitis pigmentosa without pigment. Am J Ophthalmol 1976; 81: 417-419.
Hatta M, Hayasaka S, Kato T, Kadoi C. Retrobulbar optic neuritis and rhegmatogenous retinal datachment in a fourteen years old girl with retinitis pigmentosa sine pigmento.Ophthalmologica 2000; 214: 153-155.
Weleber RG, Evans KG. Retinitis pigmentosa and allied diseases. In: Ryan SJ. Retina St Louis: Mosby; 2001; Vol I, chap 18.
Leys AM. Systemic ophthalmology. In: Duane´s ophthalmology on CD ROM. Philadelphia: JB Lippincott; 1995; Vol. 5, chap 31.
Rothova A, Ooijman F, Kerkhoff F, Van der Lelij A, Lokhorst HM. Uveitis Masquerade Syndromes. Ophthalmology 2001; 108: 386-399.
Díaz-Valle D, Miguélez R, Toledano N. Síndromes mascarada de uveítis. En: Gegúndez Fernández JA. Aproximación clínica al diagnóstico de las uveítis. Sociedad Española de Oftalmología: Madrid; 2002; 201-212.
Thirkill CE, Roth AM, Takemoto DJ, Tyler NK, Kelter JL. Antibody indications of secondary and superimposed retinal hypersensitivity in retinitis pigmentosa. Am J Ophthalmol 1991; 112: 132-137.
Gouras P. Transmision electrn microscopic observations of vitreous abnormalities in retinitis pigmentosa. Am J Ophthalmol 1987; 103: 345.