Fig. 1: Hipertrofia folicular en fondo de saco inferior y masa folicular salmón-anaranjada conjuntival bulbar superior OI.
SEMINARIO DE CASOS CLÍNICOS
GONZALO OLALLA M1, MEDIAVILLA PEÑA E1, JADRAQUE RUIZ Y2, LARRUCEA MARTÍNEZ I3, AREIZAGA OSÉS A1, SAN CRISTÓBAL EPALZA J1, PEREDA MARTÍNEZ E4
Hospital San Eloy, Baracaldo. Vizcaya.
1 Licenciado en Medicina. Residente de Oftalmología.
2 Licenciada en Medicina. Médico adjunto servicio Oftalmología.
3 Licenciado en Medicina. Jefe de servicio de Oftalmología.
4 Licenciada en Medicina. Médico adjunto servicio Anatomía
Patológica.
RESUMEN
Introducción: Los linfomas orbitarios representan un 5% del total de los linfomas no Hodgkin extranodales. Los tipo MALT son los linfomas oculo-orbitario más frecuentes. Se debe siempre descartar afectación sistémica.
Caso clínico: Presentamos el caso de un varón joven con una masa salmón-anaranjada folicular conjuntival tarsal que tras biopsia es diagnosticado de linfoma conjuntival MALT de bajo grado (estadio III A) y tratado con seis ciclos de quimioterapia R-CHOP y posterior extirpación esplénica.
Conclusión: La supervivencia global de los linfomas conjuntivales MALT es aproximadamente un 80% en 10 años aunque un 20% presentan afectación sistémica en el momento del diagnóstico. En general tienen buen pronóstico, las recurrencias suelen ser locales y rara vez metastatizan estando presente el riesgo de afectación extraocular hasta 5 años después.
Palabras clave: Linfoma conjuntival, linfoma MALT, linfoma B extranodal, Helicobacter pylori.
INTRODUCCIÓN
Los linfomas orbitarios representan un 5% del total de los linfomas no Hodgkin extranodales, siendo primarios de órbita un 25%. Los tipo MALT (tejido linfoide asociado a mucosas) son linfomas B extraganglionares de bajo grado derivados de linfocitos de la zona marginal con alteraciones moleculares que han bloqueado su apoptosis (1). Constituyen el tipo más frecuente de linfoma óculo-orbitario manifestándose la mayoría como una enfermedad localizada, siendo la forma conjuntival la menos frecuente, pudiéndose en este caso hablar de CALT por ser tejido linfoide asociado a la conjuntiva (1,2). Se debe siempre descartar que no se trate de una manifestación local de un linfoma sistémico.
Con frecuencia existen antecedentes de enfermedad autoinmune (enfermedad de Sjögren, tiroiditis de Hashimoto) o inflamatoria (gastritis por Helicobacter pylori), y se le ha relacionado con el VHC, VHS y clamidias (1,2).
CASO CLÍNICO
Varón de 43 años sin antecedentes personales ni familiares de interés y con historia de cuerpo extraño corneal izquierdo extraído hace dos años, que acude a nuestro servicio hace 14 meses, en septiembre de 2007, por molestias en ojo izquierdo (OI). Su agudeza visual lejana corregida (AVLc) es 1.0, la tensión ocular es normal y en la biomicroscopía del segmento anterior se detecta en el OI una hipertrofia folicular conjuntival en el fondo de saco inferior y a la eversión del párpado superior, una masa subconjuntival bulbar superior de color salmón-anaranjado (fig. 1) sin otros signos. El fondo de ojo no muestra hallazgos de interés así como la exploración del ojo adelfo.
![]()
Fig. 1: Hipertrofia folicular en fondo de saco inferior y masa folicular
salmón-anaranjada conjuntival bulbar superior OI.
Se inicia tratamiento con doxiciclina 100 mgrs cada 12 horas para cubrir la conjuntivis folicular crónica por Chlamydia psitacci y se decide biopsiar la lesión (fig. 2). Se obtiene un fragmento de conjuntiva bulbar superior de 17 x 5 mm que muestra una infiltración celular monótona difusa y en acúmulos constituida por linfocitos pequeños, algunos de núcleo hendido con ocasionales células de hábito plasmocitoide. Realizadas técnicas de inmunohistoquímica, la célula proliferada presenta inmunofenotipo B (CD 20 y CD 79 positivos), positividad dispersa para CD 3 y Bcl-2, negatividad para CD 23, ciclyna D1 y CD 10, y CD 5 no concluyente, por tanto compatible con un linfoma conjuntival (fig. 3).
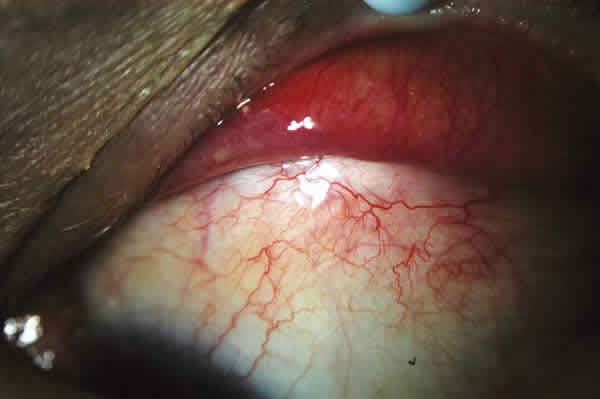
Fig. 2: Fibrosis conjuntival tras biopsia de lesión conjuntival bulbar superior
OI.
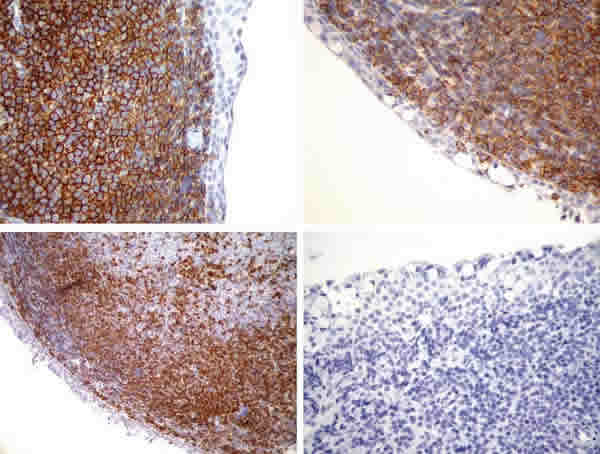
Fig. 3: IH: la célula proliferada presenta inmunofenotipo B (CD 20 y CD 79
positivos, arriba izquierda y derecha respectivamente, 40X), positividad
dispersa para Bcl-2 (abajo izquierda, 20X) y negatividad para CD 23 (abajo
derecha, 40X)
Remitimos a nuestro paciente al servicio de Oncología para estudio sistémico y tratamiento. La TAC cérvico-toraco-abdómino-pélvica muestra aumento de densidad en el espacio prevascular mediastínico sin clara masa, adenopatías perihiliares, múltiples lesiones focales hipodensas y sólidas en el bazo que aunque inespecíficas plantean la posibilidad de afectación por linfoma, así como adenopatías mesentéricas. La RNM de órbita y la punción de médula ósea resultan normales. La gammagrafía con Galio demuestra una hipercaptación en la región hiliar bilateral.
Mes y medio después el paciente es diagnosticado de linfoma MALT de bajo grado de conjuntiva en estadío III A e inicia tratamiento quimioterápico (QT) con R-CHOP: ciclofosfamida, adriamicina, vincristina, prednisona y rituximab. Es valorado por nosotros al completar el primer ciclo mostrando respuesta a nivel conjuntival desapareciendo prácticamente la lesión en el OI (fig. 4). En diciembre, completados ya tres ciclos, presenta remisión completa conjuntival (fig. 5) y TAC sin hallazgos y seis meses después del diagnóstico, finalizados los seis ciclos previstos con buena tolerancia, desaparición de las adenopatías y analítica normal, persiste la afectación esplénica aunque con PET negativo, decidiéndose la extirpación del bazo en octubre de 2008, resultando la anatomía patológica normal. El paciente se encuentra asintomático actualmente con remisión completa del proceso y sin tratamiento.
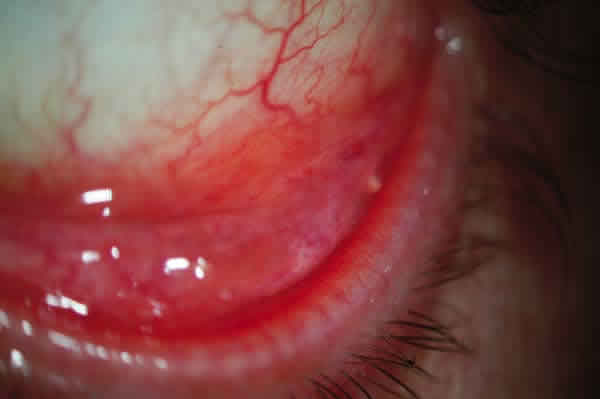
Fig. 4: Desaparición casi completa de la tumoración tras el primer ciclo de
R-CHOP.
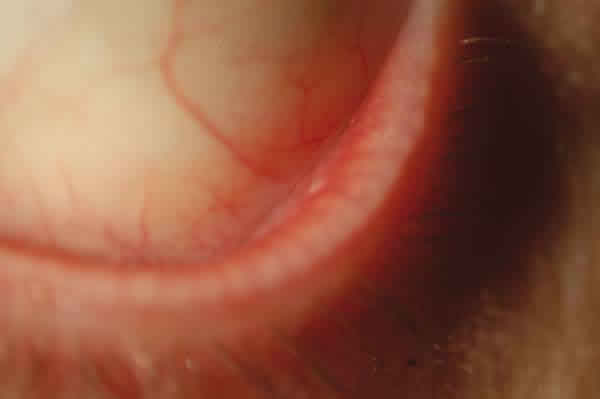
Fig. 5: Aspecto de la conjuntiva del fórnix inferior del OI tras completar los 6
ciclos de QT.
DISCUSIÓN
Las lesiones linfoproliferativas de la conjuntiva son un grupo heterogéneo de patologías que oscilan desde la hiperplasia linfoide reactiva benigna hasta los linfomas malignos (2,3). Los tipo MALT son linfomas B extraganglionares de bajo grado derivados de linfocitos de la zona marginal con alteraciones moleculares que han bloqueado su apoptosis (1). Se han descrito en tracto gastrointestinal, glándula salival, tiroides, órbita, pulmón y mama. Constituyen el tipo más frecuente de linfomas oculo-orbitarios (prevalencia que oscila desde 8-42% del total según las series) (4,5), representando la órbita su localización más habitual (2,3). Los linfomas conjuntivales se asocian a los sistémicos en un 31% de los casos, siendo los de mayor riesgo los localizados en el fórnix o en la conjuntiva bulbar interpalpebral. Los palpebrales sin embargo alcanzan el 64-70%. Por tanto es necesario un seguimiento del paciente a largo plazo puesto que un linfoma sistémico puede manifestarse años más tarde (2).
Tienen predilección por el sexo femenino (5) y son habitualmente unilaterales (65%), aunque existe un porcentaje no despreciable de bilaterales que además asocian afectación sistémica (47%) (3). La edad media de presentación son 53 años. Normalmente son asintomáticos y en algunos casos están descritas conjuntivitis previas que refuerzan la hipótesis antigénica de la patogénesis de estos linfomas. De hecho la prevalencia de Chlamydia psitacci en los CALT varía desde 0-47%, lo que lleva a algunos autores a considerar esta infección como factor de riesgo (4). La lesión más característica es una masa salmón-anaranjada en el fondo de saco no adherida a planos profundos; si se extiende a órbita puede producir ptosis o proptosis. El diagnóstico definitivo se realiza mediante la toma de una muestra generosa y un estudio inmunohistoquímico de la misma. Los cuerpos de Russel y de Dutcher generalmente indican que se trata de una lesión linfoide reactiva no neoplásica (1).
Es muy importante estadiar correctamente estos tumores (clasificación de Ann-Arbor, tabla 1) dado que aunque la mayoría se diagnostican en estadio IE (supervivencia global del 73-81% en 10 años), hasta un 20% presentan afectación sistémica en el momento del diagnóstico (5). Se han descrito múltiples alternativas terapéuticas: radioterapia local, escisión simple, crioterapia, quimioterapia sistémica con CHOP, doxicilina, IFN-a intralesional, mitomicina-C, etc (1,3). Incluso se ha descrito algún caso de regresión espontánea de la lesión tras practicar una biopsia excisional de la misma, hecho relativamente frecuente en los MALT gástricos tras erradicar la infección por Helicobacter pylori. Se especula en estos casos el papel que puede jugar el tratamiento con doxiciclina oral (5,6). La radioterapia con protección cristaliniana resulta de elección en linfomas exclusivamente orbitarios (estadio IE) con una eficacia cercana al 100% aunque son bien conocidos sus efectos secundarios (catarata, ojo seco, estenosis de puntos lagrimales, vasculopatía retiniana, neuropatía óptica, atrofia grasa orbitaria, diplopía, ) y precisa de múltiples sesiones. Actualmente el tratamiento con quimioterapia CHOP muestra porcentajes de eficacia similares (79-100%) con superioridad en el caso de afectación extraocular (5), razón por la que se administró en nuestro paciente. Entre los efectos secundarios de la quimioterapia destacan inmunodepresión (trombocitopenia y neutropenia con el consiguiente riesgo de infecciones), alteraciones genitourinarias, gastrointestinales, dermatológicas y pulmonares (5).

En cuanto al pronóstico, la positividad de los antígenos tumorales Ki-67 y p-53, los niveles altos de LDH, el tipo de linfoma y si presenta afectación sistémica guardan correlación con el grado de malignidad. En general tienen buen pronóstico, las recurrencias suelen ser locales y rara vez metastatizan aunque el riesgo de afectación extraocular está presente hasta 5 años después (1).
BIBLIOGRAFÍA
Sampedro López A, Morán M, Pinto Blázquez J, Barbón García JJ, Sánchez García FJ, Abelairas Gómez V. Linfomas oculares. Studium Ophthalmologicum 2007; vol XXV(2), 103: 111.
Yu SC, Chiu SI, Ng CS, Chan HH, Tse RK. Localized conjuntival mucosa-associated lymphoid tissue (MALT) lymphoma is amenable to local chemotherapy. Int Ophthalmol 2007; DOI 10.1007/s10792-007-9102-5.
Farmer JP, Lamba M, Merkur A, Lamba WR, Hodge WG, Jordan DR, Sengar DPS, Burns BF. Characterization of lymphoprolifertive lesions of the conjuntiva: inmunohistochemical and molecular genetic studies. Can J Ophthalmol 2006; 41(6), 753-760.
Sjö NC, Foegh P, Juhl BR, Nilsson HO, Prause JU, Ralfkiaer E, Wadström T, Heegaard S. Role of Helicobacter pylori in conjuntival mucosa-associated lymphoid tissue lymphoma. Ophtahlmology 2007; 114(1), 182-186.
Ben Simon GJ, Cheung N, McKelvie P, Fox R, McNab AA. Oral Chlrambucil for extranodal, marginal zone, B-cell lymphoma of mucosa-associated lymphoid tissue of the orbit. Ophthalmology 2006; 113(7), 1209-1213.
Matsuo T, Ichimura K, Yoshino T. Spontaneus regression of bilateral conjuntival extranodal marginal zone B-cell lymphoma of mucosa-associated lymphoid tissue. J Clin Exp Hematopathol 2007, 47(2), 79-81.