SEMINARIO DE CASOS CLÍNICOS
Displasia fibrosa esfeno-orbitaria: a propósito de un caso
SAN CRISTÓBAL EPALZA J1, GONZALO OLALLA ML2, MEDIAVILLA PEÑA E1, AREIZAGA OSÉS A1, IBÁÑEZ AGUIRRE JM3, LARRUCEA MARTÍNEZ I4
Servicio de Oftalmología. Hospital San Eloy.
Vizcaya. España.
1 Residente
de Oftalmología. E-mail:
jscepalza@gmail.com
2 Médico
Adjunto del Servicio de Oftalmología.
3 Jefe de
Sección de la Unidad de Retina del Servicio de Oftalmología.
4 Jefe de
Servicio.
RESUMEN
Introducción: La displasia fibrosa esfenoidal es una enfermedad muy poco frecuente que progresa paulatinamente pudiendo provocar la compresión de estructuras como el nervio óptico. Cuando esto ocurre, puede ser necesaria una descompresión quirúrgica para evitar un daño irreversible del nervio óptico.
Caso clínico: Mujer de 31 años con displasia fibrosa esfenoidal bilateral, que presentó un cuadro compresivo del nervio óptico derecho que requirió intervención quirúrgica y generó secuelas en la función visual de dicho ojo. Asimismo, presentó 3 episodios de visión borrosa y alteración de la percepción del color en ojo izquierdo con disminución de la agudeza visual, que recuperó totalmente tras tratamiento con corticoides orales. En la actualidad la función visual del ojo izquierdo se conserva, y hasta el momento no se ha requerido intervención quirúrgica en dicho ojo.
Palabras clave: Displasia fibrosa, esfenoides, neurofibromatosis.
INTRODUCCIÓN
La displasia fibrosa es una enfermedad ósea que aparece normalmente antes de la tercera década de la vida. Se habla de displasia monostótica si afecta únicamente a un hueso, poliostótica si implica la afectación de varios huesos, y síndrome de McCune-Albright cuando además asocia hiperpigmentación cutánea y pubertad precoz. La etiología es desconocida, aunque en el síndrome de McCune-Albright se ha descubierto una asociación con un estado de mosaico para una mutación en el gen de la proteína Gsa (GNAS1), situado en el cromosoma 20q.
CASO CLÍNICO
Mujer que acudió por primera vez a nuestro servicio en septiembre de 1996 por presentar reducción del campo visual y de la agudeza lejana y cercana (AVL, AVC) de un año de evolución, en ojo derecho (OD). No presentaba antecedentes oftalmológicos de interés y entre los antecedentes sistémicos sólo era reseñable una anemia ferropénica por menorragias. Presentaba antecedentes familiares de neurofibromatosis tipo 1.
En la exploración presentaba una AVL por OD de 0,3 mejor corregida y de 1,0 por el ojo izquierdo (OI). Se observaba un defecto pupilar aferente relativo en el OD. La biomicroscopía del segmento anterior fue normal en ambos ojos (AO). En el fondo del OD se apreciaba un borramiento del borde nasal de la papila, siendo el polo posterior normal así como la exploración funduscópica del OI.
Se le realiza una campimetría Humphrey, programas 30-2 Umbral y Sita Standard, que objetivó en el OD un defecto casi absoluto con un pequeño islote nasal superior respetado. El campo visual del OI fue normal. El electrorretinograma no mostró alteraciones patológicas en ninguno de los dos ojos. Los potenciales evocados visuales fueron patológicos en el OD, demostrando menor amplitud y latencias retrasadas con respecto a los valores normales. Los potenciales del OI fueron normales. La angiografía fluoresceínica fue normal en AO.
Se solicitó una resonancia nuclear magnética (RNM) cerebral, en la que se describió un patrón en vidrio esmerilado difuso en la región esfenoidal, afectando al cuerpo y a las alas mayores, que provocaba un estrechamiento bilateral de los canales ópticos, mayor en el lado derecho (fig. 1).

Fig. 1: RNM en la que se aprecia el patrón en vidrio esmerilado en región
esfenoidal, con estrechamiento de ambos canales ópticos, más importante en el
derecho.
Se le practicó una craniectomía frontal derecha con fresado del techo de la órbita para descomprimir el nervio óptico por parte de Neurocirugía.
Dos meses después, la paciente vuelve a acudir por un episodio urgente de dolor ocular derecho con limitación moderada de todos los movimientos oculares extrínsecos, más acusada en la elevación y en la abducción, compatible con un posible síndrome de hendidura. A raíz de este episodio, es derivada nuevamente a Neurocirugía, quienes realizan una segunda intervención por nuevo crecimiento de la lesión ósea (apófisis clinoides).
A partir de este momento la paciente ha mantenido con una situación estable en la que la AVL por el OD es de 0,4 con mucha dificultad por su campo visual nasal, y por el OI ve 1,0. La papila del OD es atrófica, siendo el fondo del OI normal. En el campo visual del OD quedó un defecto casi absoluto con zona nasal superior menos afectada (fig. 2), mientras que el campo del OI está conservado.
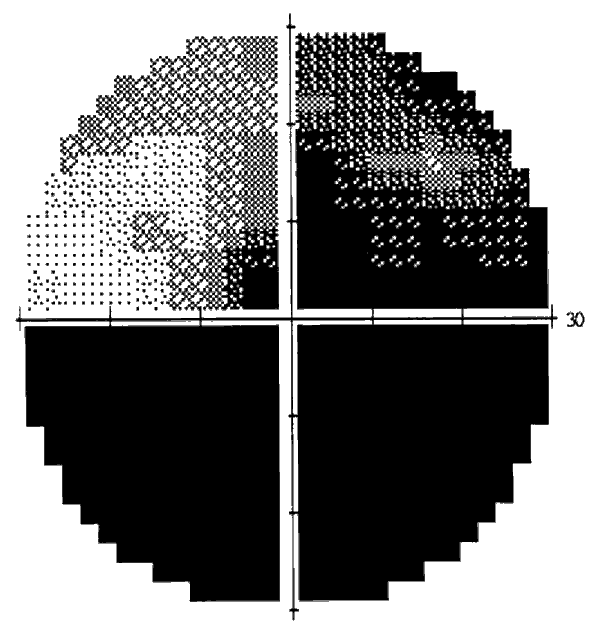
Fig. 2: Campimetría del OD después de la segunda intervención quirúrgica, en el
que se observa el defecto total, con islote nasal superior residual.
Después de 10 años de evolución comienza con episodios de pérdida de AVL por el OI con alteración de la percepción del color, sin dolor.
Durante los 3 episodios que ha tenido hasta el momento, se objetiva una disminución de la AV, llegando a reducirse en uno de los brotes hasta 0,2, con recuperación hasta 1,0 tras el tratamiento con corticoides orales a cargo de Neurocirugía. La motilidad ocular y la biomicroscopía no muestran alteraciones. En el fondo del OI se aprecia un borramiento de la papila durante los episodios, que vuelve a la normalidad con la remisión del cuadro. La exploración del OD es estable.
Al realizar la campimetría, se observa en el OI un escotoma relativo central durante los cuadros (fig. 3), que también cede con el tratamiento.
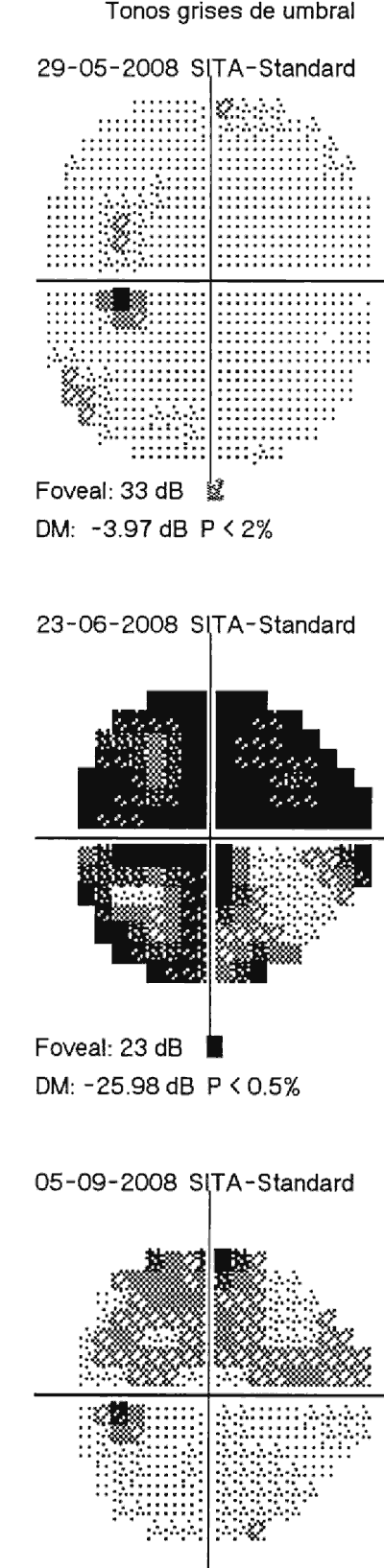
Fig. 3: Campimetría del OI en diferentes fases del episodio actual, mostrando
según la actividad del cuadro en ese momento distintos grados de afectación.
Se solicitó una nueva RNM cerebral y se derivó a Neurocirugía para valoración de nueva intervención descompresiva, en esta ocasión del lado izquierdo. No se ha considerado realizar cirugía por el momento y la paciente se encuentra actualmente asintomática desde hace un año, con controles oftalmológicos cada 6 meses en los que no se ha objetivado progresión de la compresión del nervio óptico.
DISCUSIÓN
Con los datos vistos hasta ahora, los diagnósticos diferenciales que nos planteamos fueron la neurofibromatosis tipo 1 (NF-1) y la displasia fibrosa.
Debido a los antecedentes familiares que presentaba nuestra paciente, el primer diagnóstico de sospecha fue la neurofibromatosis tipo 1 (NF-1), que asocia en un 10% displasia unilateral del esfenoides y que puede constituir una de las primeras manifestaciones clínicas de dicha enfermedad (1). La paciente debía cumplir 2 de los 7 criterios diagnósticos de NF-1 (tabla 1) para que el cuadro fuera catalogado como tal. En nuestro caso esos 2 criterios serían una lesión ósea característica (como la displasia unilateral del esfenoides, o la displasia o el adelgazamiento cortical de huesos largos), y tener un pariente de primer grado afectado de NF-1. Sin embargo, nuestra paciente presenta una displasia bilateral y además se le realizaron dos estudios genéticos diferentes en los que se afirma que no es portadora de la mutación del haplotipo paterno responsable de la NF-1. A pesar de no ser portadora de dicha mutación, más del 50% de los casos de neurofibromatosis tipo 1 corresponden a mutaciones de novo (2). Si bien no tenemos pruebas concluyentes de la relación de la displasia fibrosa de esfenoides con la NF-1 en esta paciente, la coincidencia de ambos procesos en su familia puede parecer algo más que casual.

La siguiente posibilidad diagnóstica era la displasia fibrosa, que en este caso sería monostótica porque afecta únicamente al esfenoides. La afectación esfenoidal es la segunda en frecuencia (3), por detrás del hueso frontal. En algunas series, la afectación del hueso maxilar también es más frecuente que la del esfenoides (4). Otras manifestaciones que pueden aparecer en la displasia fibrosa esfenoidal son la asimetría facial, proptosis, desplazamiento del globo ocular, diplopia, parálisis de nervios craneales, maloclusión, hipertensión intracraneal, obstrucción nasal o de conductos nasolagrimales, o incluso dolor orbitario o cefalea difusa.
Este tipo de lesiones solitarias podrían plantear un diagnóstico diferencial con el fibroma oseificante (5); sin embargo, el propio patrón que se aprecia en la resonancia magnética nos orienta hacia una displasia fibrosa.
El proceso displásico produjo un proceso compresivo en el nervio óptico del OD, que ha dejado las secuelas mencionadas durante la exposición del caso clínico. En el OI, sospechábamos que se estaba empezando a producir también una compresión del nervio óptico; sin embargo, la RNM no detectó una progresión de la displasia que nos confirmara la compresión. Por lo tanto, nuestra hipótesis gira en torno a un cuadro irritativo-compresivo que provoca la inflamación del nervio óptico, remitiendo con tratamiento médico.
En caso de confirmarse el diagnóstico de la neuropatía óptica compresiva, el tratamiento consiste en corticoides orales (prednisona) en dosis de 1 mg/kg/día durante 1 mes, procediendo posteriormente a realizar una pauta descendente hasta lograr la dosis mínima que mantiene a la paciente estable. Si este tratamiento fracasa, la siguiente opción consistiría en la cirugía descompresiva (a cargo de Neurocirugía). Se puede optar por hacer un simple curetaje, o plantear una intervención más agresiva con resección del hueso displásico con o sin reconstrucción.
Sigue sin confirmarse cuál es la mejor actitud a seguir en estos casos. La mayoría de los autores desaconsejan descomprimir el nervio de forma profiláctica, ya que este tipo de cirugía produce invariablemente una lesión del nervio óptico, por lo que el problema actual es saber hasta qué punto se debe dejar que se dañe el nervio óptico. Si se realiza la intervención quirúrgica demasiado pronto, corremos el riesgo de dañar el nervio óptico durante la cirugía; en cambio, si esperamos demasiado tiempo, el nervio óptico izquierdo podría sufrir daños irreversibles debido a la compresión (4).
CONCLUSIONES
La displasia fibrosa esfenoidal puede provocar la compresión de los nervios ópticos, pudiendo provocar una importante pérdida visual del lado afectado.
El tratamiento durante los brotes compresivos consiste en corticoides orales.
El tratamiento definitivo es la cirugía descompresiva.
BIBLIOGRAFÍA
-
Romero C, Bertrés P, Vidondo ME, Ghisi JP, Mazzucco J, Ternak A. Displasia esfeno-orbitaria unilateral. Malformación poco frecuente asociada a neurofibromatosis tipo 1: Hallazgos en RM. www.diagnosticojournal.com
-
Puig Sanz L. Síndromes neurocutáneos. www. aeped.es/protocolos/dermatologia
-
Katz BJ, Nerad JA. Ophthalmic manifestations of fibrous dysplasia. Ophthalmology 1998; 105: 2207-2215.
-
Lee JS, FitzGibbon E, Butman JA, Dufresne CR, Kushner H, Wientroub S, Robey PG, Collins MT. Normal vision despite narrowing of the optic canal in fibrous dysplasia. New England Journal of Medicine 2002; 347(21): 1670-1676.
-
Alba García JR, Armengot Carceller M, Pérez Fernández, CA, Díaz Fernández A, Taleb C Campos Catalá A, Basterra Alegría J. Una forma excepcional de fibrosa craneofacial: displasia fibrosa de cornete medio. Acta Otorrinolaringol Esp 2002; 53: 291-4.