SEMINARIO DE CASOS CLÍNICOS
Retraso diagnóstico de glioma óptico en un caso de alta miopía
Reche-Sainz JA1, Gutiérrez-Montero O1, Domingo-Gordo B2
Servicio de Oftalmología. Hospital
Universitario de Fuenlabrada. Madrid.
1
Licenciado en Medicina. E-mail:
jalbres@hotmail.com
2 Doctor en
Medicina.
RESUMEN
Introducción: El glioma óptico es el tumor primario más frecuente del nervio óptico, habitualmente asociado a la neurofibromatosis tipo I. Suele tener un crecimiento insidioso, y se manifiesta por un descenso lento, progresivo e indoloro de la visión del ojo afecto.
Caso clínico: Mujer 20 años, con antecedente de cuadro ansioso-depresivo, fue remitida por disminución de visión del ojo izquierdo de lenta evolución. La agudeza visual del ojo derecho era de 1,0 (–4 –0,75 a 15º), y en el ojo izquierdo de 0,05 (–13,5 –4 a 5º). En la exploración, sólo era reseñable una coroidopatía miópica severa del ojo izquierdo. Se le diagnosticó de ambliopía por anisometropía. A los 6 meses, la paciente presentó un cuadro autolimitado de alucinaciones auditivas complejas que se consideraron atípicas en su contexto psiquiátrico. Para descartar un origen orgánico de estos síntomas, se le realizó una prueba de neuroimagen que incidentalmente mostró una tumoración de nervio óptico izquierdo compatible con glioma óptico.
Palabras clave: Glioma óptico, alta miopía.
INTRODUCCIÓN
El glioma óptico es el tumor primario más frecuente de la vía óptica anterior. Aparece más habitualmente en la edad pediátrica, su crecimiento suele ser lento e insidioso, y muchas veces clínicamente silente. Se expone el caso de una paciente que presentó un glioma óptico en un ojo miope magno, cuyo diagnóstico se retrasó al estar enmascarado por una ambliopía por anisometropía.
CASO CLÍNICO
Una mujer de 20 años, con antecedentes de depresión mayor y ansiedad generalizada, acudió a consulta por pérdida de visión del ojo izquierdo (OI). La paciente refería que si bien la agudeza visual (AV) del OI había sido siempre muy baja, en los últimos meses notaba cierto empeoramiento visual. En la exploración, la AV del ojo derecho (OD) era de 1,0 (–4 –0,75 a 10º) y la del OI, 0,05 (–13,5 –4 a 5º). La exploración del segmento anterior sólo reveló una mínima opacidad subcapsular cristaliniana del OI. Funduscópicamente, se observaba una gran retinocoroidosis miopica en el OI. Se le diagnosticó de ambliopía profunda OI por anisometropía agravada por una catarata subcapsular incipiente en ese ojo. Se le remitió a Atención Primaria con la recomendación de revisiones oftalmológicas periódicas. Sin embargo, a los 4 meses, la paciente experimentó un cuadro autolimitado de alucinaciones auditivas («oía discusiones» y «voces que comentaban sus pensamientos»), por lo que fue valorada psiconeurológicamente. Al tratarse de síntomas atípicos de un trastorno afectivo, y para descartar un origen orgánico, se le realizó una resonancia magnética cerebral que evidenció la existencia de un engrosamiento de nervio óptico izquierdo localizado en su porción más posterior y en la región adyacente del quiasma óptico, muy sugestivo de glioma óptico (figs. 1 y 2). Reevaluada oftalmológicamente, la AV mejor corregida del OI se mantenía en 0,05, y la papila óptica del OI tenía un aspecto ligeramente pálido, difícil de valorar por las alteraciones miópicas (fig. 3). Campimétricamente, se objetivó una depresión generalizada del OI sin afectación del OD. La exploración neurológica no aportó ningún otro hallazgo, y en la dermatológica no se encontró ningún signo de neurofibromatosis tipo 1 (NF-1).
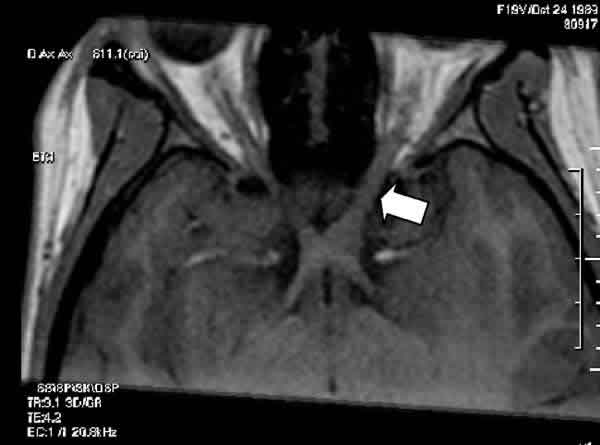
Fig. 1: En esta reconstrucción transversal en modo T1, se observa el
engrosamiento del nervio óptico izquierdo (flecha) en la vecindad del quiasma.
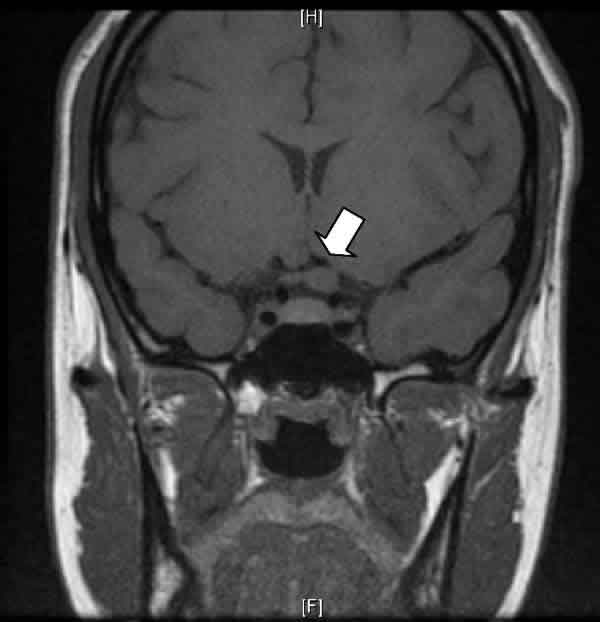
Fig. 2: En este corte coronal en modo T1, se objetiva el agrandamiento de la
sección del nervio óptico izquierdo (flecha).

Fig. 3: Aspecto de la papila óptica del ojo afecto.
DISCUSIÓN
Los gliomas de la vía óptica son neoplasias primarias que afectan principalmente al nervio y quiasma óptico. Representan el 2-5% de los tumores cerebrales en la edad pediátrica (1,2). Aparecen habitualmente antes de los 10 años de edad (59-70%), siendo mucho menos frecuentes en la edad adulta y de pronóstico más desfavorable (2). La localización más habitual es el nervio óptico, pero pueden afectar también al quiasma y extenderse a estructuras contiguas como el hipotálamo y el tercer ventrículo (3).
Existe una fuerte asociación del glioma óptico a la NF-I, aunque la frecuencia descrita es variable, 10-70% según las diferentes series (4). Por otro lado, se ha estimado que hasta un 15% de pacientes con NF-1 desarrollan un glioma óptico (2). Estos gliomas asociados a NF-1 suelen tener una evolución benigna, pero con mayor frecuencia son multifocales, bilaterales y de localización retroquiasmática (2).
Los gliomas ópticos clínicamente pueden permanecer asintomáticos durante largo tiempo, o manifestarse por una pérdida visual lenta e indolora que se puede acompañar de edema o atrofia papilar (1,2). La proptosis y estrabismo aparecen en los casos de ocupación orbitaria importante (2,3). En el caso aquí expuesto, la pérdida visual estaba enmascarada por baja visión producida por la alta miopía del ojo afecto. La ausencia de otras manifestaciones no hacía sospechar la existencia de una lesión del nervio óptico.
En el caso de los gliomas quiasmáticos, la pérdida visual es también lenta e insidiosa pero con carácter bilateral con afectación campimétrica bitemporal. Menos habitualmente, pueden producirse alteraciones endocrinas como pubertad precoz, alteraciones hipotalámicas, spasmus nutans e hidrocefalia obstructiva en los casos de invasión supraselar (2). En el caso expuesto las alucinaciones auditivas que la paciente experimentó, parece que guardaban más relación con su cuadro psiquiátrico, que con la existencia del glioma óptico.
Histológicamente, la gran mayoría son astrocitomas pilocíticos, que son tumoraciones de bajo grado de crecimiento generalmente intraneural, aunque con potencial invasivo y de malignización (5).
El diagnóstico se confirma mediante pruebas de neuroimagen preferiblemente con resonancia magnética cerebral (2,3). Ésta permite una evaluación más precisa de los nervios ópticos y quiasma, así como de cintillas e hipotálamo. Las lesiones característicamente son isointensas o hipointensas en modo T1, e hiperintensas en T2 y se realzan homogéneamente con galodinio (1-4).
La evolución espontánea de estos tumores suele ser lenta e insidiosa, pero muchas veces es impredecible. Se han descritos casos clínicamente similares, pero con evoluciones muy dispares: estabilización, progresión e incluso regresión tumoral espontánea (2).
Se ha comprobado que la única afectación del nervio óptico y la presencia de NF-1 son factores de buen pronóstico, mientras que una edad menor de 1 año en la presentación lo empeora (2,4).
El tratamiento no está estandarizado y ha de ser considerado en función de las manifestaciones clínicas y de la progresión radiológica del tumor (1,2). En general, de no existir afectación clínica significativa, se plantea una actitud expectante con al menos una prueba de neuroimagen anual. Si apareciese pérdida visual severa (AV <0,5), o se demostrase progresión en la neuroimagen, se suele plantear quimioterapia como tratamiento de elección, sobre todo en niños menores de 5 años (2,4). La radioterapia tiene muchos efectos secundarios en estos pacientes (2,4), por lo que sólo se indicaría en caso de fracaso de la quimioterapia, y en pacientes de más edad (1,2,4). La cirugía tiene un papel controvertido, pues puede acelerar la pérdida visual (1-4). Se plantea para realización de biopsias en casos atípicos, en casos de gran ocupación orbitaria con ojos de muy baja visión, y en las hidrocefalias obstructivas (2).
Consultado el Servicio de Neurocirugía, en nuestro caso se optó por la observación clínica y la monitorización del posible crecimiento tumoral mediante neuroimagen en plazos de 6 meses.
Como conclusión, este caso demuestra la posibilidad de que el diagnóstico de glioma óptico se haga tardíamente. Inicialmente no hubo sospecha diagnóstica de glioma óptico, ya que se trataba de un ojo miope magno y ambliope y sin más manifestaciones visuales o sistémicas (NF-1).
BIBLIOGRAFÍA
-
Sylvester CL, Drohan LA, Sergott RC. Optic-nerve gliomas, chiasmal gliomas and neurofibromatosis type1. Curr Opin Ophthalmol 2006; 17: 7-11.
-
Liu GT. Optic gliomas of anterior visual pathway. Curr Opin Opthalmol 2006; 17: 427-431.
-
Binning MJ, Liu JK, Kestle JR, Brockmeyer DL, Walker ML. Optic nerve gliomas: a review. Neurosurg Focus 2007; 23: 1-8.
-
Wilhelm H. Primary optic nerve tumours. Curr Opin Ophthalmol 2009; 22: 11-18.
-
Shamji MF, Benoit BG. Syndromic and sporadic pediatric optic pathway gliomas; review of clinical and histopathological differences and treatment implications. Neurosurg Focus 2007; 23: 1-9.