SEMINARIO DE CASOS CLÍNICOS
Retinosis pigmentosa y sordera: un reto diagnóstico para el oftalmólogo
RODRÍGUEZ CALVO DE MORA M1, RODRÍGUEZ MORENO G1, ESPAÑA CONTRERAS M1
Hospital Regional
Universitario Carlos Haya. Málaga.
1
Licenciado en Medicina.
RESUMEN
Introducción: Existen múltiples síndromes que presentan trastornos auditivos y alteraciones oftalmológicas. El Síndrome de Usher (USH) es un desorden hereditario autosómico recesivo que implica hipoacusia neurosensorial y retinosis pigmentosa.
Caso clínico: Se presentan los casos de dos hermanas no univitelinas de progenitores no cosanguíneos que muestran hipoacusia desde el nacimiento.
En la paciente 1 a los 16 años se observa una disminución de su mejor agudeza visual corregida (MAVC). En sucesivas exploraciones se aprecian osteoclastos periféricos y cuerpos hialinos exofíticos peripapilares, autofluorescentes, típicos del Síndrome de Usher. En la paciente 2 se observaron alteraciones periféricas típicas de retinosis pigmentaria a una edad más temprana, campos visuales con constricción de campo visual y depósitos prepapilares similares a los de su hermana.
Palabras clave: Retinosis pigmentosa, síndrome de Usher.
INTRODUCCIÓN
El Síndrome de Usher es un desorden hereditario autosómico recesivo que implica hipoacusia neurosensorial y retinosis pigmentosa (RP). Representa la causa más frecuente de sordoceguera infantil de causa genética (50% en Estados Unidos) y muestra una gran heterogeneidad clínica y genética (1).
CASO CLÍNICO
Se presentan los casos de dos hermanas no univitelinas de progenitores no cosanguíneos sin antecedentes familiares positivos que muestran hipoacusia desde el nacimiento y que siguieron revisiones oftalmológicas periódicas como screening.
La paciente 1 presentaba a los 7 años visión de unidad, hipermetropía de + 9 dioptrías en ambos ojos, con ortoforia y sin supresión de ningún ojo. Las exploraciones oftalmológicas rutinarias no mostraron alteraciones significativas, salvo un leve descenso de la agudeza visual mejor corregida (AVMC) medida por optotipo de Snellen de 2/3 a los 14 años. Sin embargo, a los 16 años se observa una disminución de AVMC a 1/2, una opacidad cristaliniana subcapsular posterior, algunas alteraciones periféricas sine pigmento y osteoclastos y una leve atrofia macular en ojo de buey en el ojo izquierdo (OI).
Ante la presencia de retinitis pigmentosa y sordera se procedió a hacer diagnóstico diferencial entre los distintos síndromes que pueden asociar retinosis pigmentaria y sordera. Se descartaron todos ellos, ya que la paciente no mostraba otros signos oftalmológicos ni sistémicos de dichas patologías: Cockayne (enanismo, retraso mental, calcificaciones intracraneales), Bardet-Bield (obesidad, braquidactilia, polidactilia, hipogenitalismo y nefropatía), Flynn-Aird (ataxia, epilepsia, demencia, neuritis periférica, ulceración cutánea, calvicie, cambio óseos), Kearns-Sayre (ptosis, oftalmoplejía externa, arritmia, alteraciones del sistema nervioso central, talla baja, diabetes), Enfermedad de Almström (miocardiopatía, Diabetes mellitus, obesidad, acantosis nigricans, calvicie, hipogenitalismo, hipertrigliceridemia) y ataxia de Friedrich (ataxia espinocerebelosa, miocardiopatía y diabetes) (1). El electrorretinograma de la paciente mostraba abolición de las respuestas de los bastones y reducción de las respuestas fotópicas. La paciente fue diagnosticada de Síndrome de Usher a falta de confirmación genética.
En evolución posterior de la paciente se constató en la funduscopia además de algunos osteoclastos periféricos, dos grupos de lesiones nodulares papilares blanquecinas, correspondientes a cuerpos hialinos exofíticos peripapilares típicos del Síndrome de Usher (fig. 1). Se discute si estas excrecencias globulares son drusas de localización anómala por transporte axoplásmico aberrante o hamartomas astrocíticos (2,3). Las lesiones presentaban autoflorescencia al realizar retinografía con luz aneritra (fig. 2). En la actualidad la paciente mantiene una agudeza visual de 1/3.
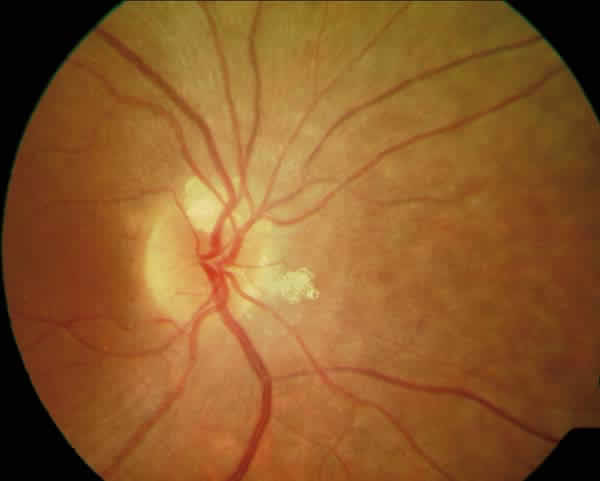
Fig. 1: Cuerpos hialinos papilares.

Fig. 2: Autofluorescencia.
La paciente 2 presentaba a la edad de 2 años (cinco años de diferencia de edad con su hermana) hipermetropía de +9,50 dioptrías y endotropía en su OI, siendo el ojo derecho (OD) dominante. A los 16 años presentaba ortoforia con AVMC de unidad en OD y de 2/3 en OI, sin alteraciones del polo anterior ni del fondo de ojo. Dado los antecedentes de su hermana, fue derivada para despistaje de Síndrome de Usher. Las pruebas neurofisiológicas eran orientativas de Síndrome de Usher.
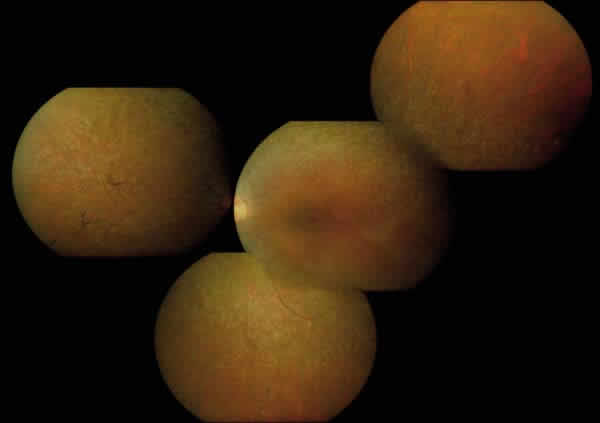
Fig. 3: Aspecto funduscópico de la paciente 1.
En sucesivas exploraciones oftalmológicas se observaron alteraciones periféricas típicas de retinosis pigmentaria, campos visuales con lesiones características de constricción de campo visual y depósitos prepapilares similares a los de su hermana (fig. 4).
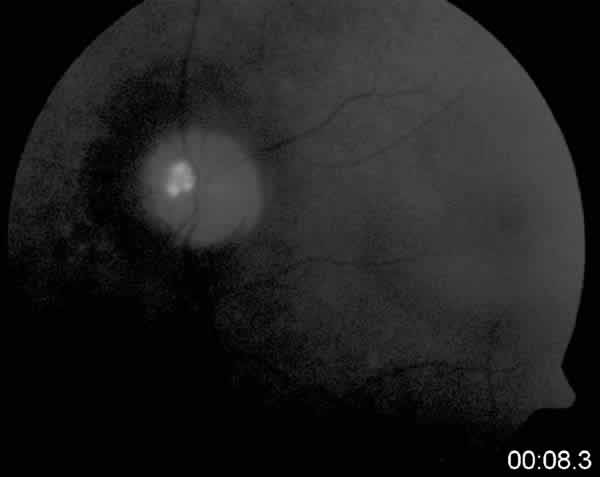
Fig. 4: Autofluorescencia.
DISCUSIÓN
El síndrome de Usher (USH) comprende una sordera autosómica recesiva con retinopatía indistinguible de la producida por la Retinosis Pigmentosa (RP). Aunque fue descrito por primera vez por Von Graefe en 3 hermanos en 1858, recibió el nombre de Carles Usher porque fue quien describió en 1914 su carácter hereditario y lo definió como una entidad independiente (4).
El USH es el que más frecuentemente se asocia a la RP, con una prevalencia aproximada del 18%. En Estados Unidos es el responsable del 50 a 66% de los casos de personas ciegas y sordas, según las series (1,5). En Estados Unidos se ha calculado que la prevalencia de USH es de 1,8 a 6,2 casos por 100.000 (1). En nuestra población la prevalencia del síndrome de Usher se ha estimado en un 4,2/100.000, aunque recientemente estas estimaciones están en revisión y se considera que la prevalencia de la enfermedad podría llegar incluso al triple (5).
Existen múltiples síndromes que presentan trastornos auditivos y alteraciones oftalmológicas: Treacher-Collins, Alport, Neurolúes, Cogan, Síndrome de Queratitis-Ictiosis-Sordera, Síndrome de Wolfram e infecciones neonatales por citomegalovirus y toxoplasma. Los Síndromes de Usher, Kearns-Sayre, Refsum, Cockayne, Bardet-Biedl, Almström, Flynn-Aird y la ataxia de Friedrich pueden asociar sordera con retinopatía pigmentaria (4).
El síndrome de Usher posee gran variabilidad genética y clínica. Se distinguen tres grupos. El Usher tipo I (USH1) asocia sordera neurosensitiva congénita profunda, síntomas vestibulares y retinopatía en la infancia. El tipo II (USH2) presenta sordera parcial congénita sin afectación vestibular y retinopatía más tardía. Supone el grupo más frecuente [con proporciones que varían del 67, 47 y 65% según los estudios (1,6)]. El Usher tipo III (USH3) tiene una baja frecuencia de presentación, siendo típico de los judíos asquenacíes y la población finesa (la proporción de USH3 en estos grupos poblacionales asciende al 40%). Se caracteriza por una sordera progresiva, postlingual, con respuestas vestibulares variables y retinopatía de aparición tardía (1,4). La prevalencia en España del USH2 es aproximadamente el doble del USH2, siendo el USH3 una forma muy poco frecuente en nuestro país (5).
Los pacientes con USH1 tienen nictalopía más precoz y afectación macular más temprana que los de tipo II. El 70% de los pacientes con Síndrome de Usher tipo I poseía una MAVC de 20/40 a los 29 años, frente al 94% del tipo 2. No se observó diferencia en la prevalencia de cataratas subcapsulares posteriores (1).
Desde que se descubrió el primer gen relacionado con el Síndrome de Usher en 1995 (MYO7A, causante del subtipo IB del USH1), se han identificado 11 loci y 9 genes relacionados con los distintos subtipos clínicos de Síndrome de Usher. Existen familias con ligamientos genéticos aún no descritos (7).
El análisis molecular del ligamiento genética ha demostrado que los tres subtipos del síndrome de Usher no sólo se distinguen genéticamente, sino que también existe heterogeneidad genética dentro de cada uno de ellos, de manera que cada tipo clínico del Síndrome de Usher se subdivide en varios subtipos genéticos (1). Existen al menos siete loci asociados al USH1, desde el USH1A al USH1G y se han aislado los genes responsables: El USH1A, localizado en 14q32; el subtipo más grave y frecuente de SUH, producido por el gen USH1B, se ha relacionado con el gen MYO7A, sito en el cromosoma 11q13.5, que codifica la miosina VIIA, que se encuentra en los cilios del oído interno y en los fotorreceptores y parece desempeñar una función fundamental en el metabolismo y en el transporte intracelular; el USH1C (11p14) codifica la harmonina, que actúa en el ensamblado de proteínas de adhesión celular; el USH1D (10q22) codifica la protocadherina 23, una proteína de adhesión intercelular; el USH1E (21q21); el USH1F (10q21-22) codifica para la protocadherina 15, esencial para la homeostasis de los estereocilios del oído interno y de los fotorreceptores); el gen USH1G, localizado en el cromosoma 17q24-25, que participa en soporte y que se expresa en la retina y en el oído interno. La mayoría de estos genes participan en casos de sordera no asociada a RP. Se han descrito tres loci para el USH2: 1q41 para el USH2A, que codifica la proteína denominada usherina, que también está implicado en la RP aislada; 3p23-24 para el USH2B; y el gen VLGR1, localizado en 5q14.3-q21.3, para el USH2C. La enfermedad tipo 3 se ha relacionado con el gen clarina-1, sito en el cromosoma 3q21-q25. Se ha propuesto un segundo locus para el tipo 3 en 20q, pero aún no se ha validado (1,5).
Tres de los genes relacionados con el USH1, USH1C, USH1D y USH1F presentan relación genotipo-fenotipo en sus mutaciones. Se ha descrito, sin embargo, el caso de dos gemelas monozigóticas que poseían los mismos alelos alterados en los mismos genes (USH2A) y mostraban distintos fenotipos. Este hecho sugiere que la influencia ambiental puede tener importancia en la gravedad de la enfermedad (8).
Los distintos genes alterados en los subtipos de Síndrome de Usher codifican para distintas proteínas estructurales (miosina VIIa, cadherinas, proteínas de transmembrana, glicoproteínas…), algunas de las cuales se encuentran presentes en las estructuras ciliadas de las células tanto del oído interno como de los fotorreceptores.
La localización de las proteínas relacionadas con el Síndrome de Usher en las áreas de sinapsis y periciliares de las células neurosensoriales oculares y del oído y la identificación de una red de interacciones entre dichas proteínas ha llevado a postular «El interactoma proteico del Síndrome de Usher» (El interactoma describe cómo como interactúan las proteínas para otorgar funcionalidad y sincronización de los múltiples procesos de la célula). Dicha interacción proteica explicaría los efectos pleiotrópicos de los genes alterados en el Síndrome de Usher (8).
Las excrecencias globulares parecidas a drusas en el nervio óptico y la retina adyacente típicas del Síndrome de Usher muestran crecimiento e histológicamente constituyen cuerpos hialinos idénticos a las drusas. Tienen tendencia a ser bilaterales, múltiples y parapapilares. Por tanto, a tenor de las últimas publicaciones se trata de drusas de localización atípica y no de hamartomas astrocíticos, a pesar de que se pueden situar por encima de los vasos papilares (2,3).
Las actuales terapias dependen del órgano afectado; así para la audición el implante coclear se está mostrando como una terapia interesante, especialmente en niños menores de 3 años, en los que con un apoyo adecuado se alcanzan ciertas habilidades en el habla (4,5). Para la retinosis pigmentaria una refracción adecuada, la extracción de las cataratas, el tratamiento del edema macular, prótesis para baja visión y el tratamiento con suplementos de vitamina A (15.000 UI/día de retinil palmitato) en adultos (vigilando la función hepática, la aparición de osteoporosis y evitando su uso en mujeres fértiles) pueden ayudar a mejorar la calidad de vida de los pacientes. Medicamentos con posibles efectos negativos sobre la enfermedad son la Isotretinoína y el Sildenafilo (1).
En un futuro la terapia génica, la terapia mediante células madre, la tecnología de células encapsuladas o tratamientos que impliquen la suma de estas tres podrían suponer el tratamiento definitivo contra la enfermedad, sin embargo, estas terapias tan prometedoras se deben tomar con mucha cautela (1).
CONCLUSIONES
Los niños con problemas de audición se apoyan especialmente en la visión para desarrollar sus habilidades de comunicación e independencia. Cualquier alteración oftalmológica puede afectar negativamente a este proceso, sobretodo si pasa desapercibida en los primeros años de vida. Los individuos afectos por sordoceguera requieren múltiples adaptaciones de su entorno, así como servicios de intervención educativa y sociopsicológica. Las estrategias rehabilitadoras específicas no se apoyan en la visión (logoterapia óptima) (1).
Debido a que tanto la RP como el USH implican una enfermedad genética, es necesario la realización de un árbol genético completo y es recomendable la exploración de otros miembros de la familia. Una vez establecido el diagnóstico, el consejo genético familiar es fundamental, brindando apoyo e información al paciente y su entorno (gravedad de la minusvalía visual y las dificultades que ello acarrea en la actividad cotidiana del paciente) y planteando la planificación familiar. Dado que el USH es una enfermedad autosómica recesiva, cada hijo tiene un 25% de probabilidades de resultar afectado, independientemente del número de hijos que ya muestren la enfermedad. Debe indagarse en la historia de consanguinidad (1).
La estrategia actual se basa en realizar un screening sistemático de problemas oftalmológicos (y neurofisiológico en casos seleccionados) en los niños con discapacidad auditiva y/o disfunción vestibular. Los padres de los niños afectos de USH1 suelen identificar el problema por la tardanza del infante en caminar o por la ceguera nocturna. Es fundamental conocer los síndromes que asocian alteraciones auditivas y visuales.
En los casos que se presentan se llevó a cabo un seguimiento oftalmológico anual. La aparición de lesiones características en la exploración funduscópica motivó la realización de pruebas complementarias neurofisiológicas (ERG) para la obtención de una orientación diagnóstica, la de Síndrome de Usher, en espera de confirmación genética.
BIBLIOGRAFÍA
-
Weleber RG, Gregory?Evans K. Retinosis pigmentosa y trastornos relacionados. En: Ryan SJ, Hinton DR, Schachat AP, editores. Retina. 4 ed. New York: Elsevier; 2006. p: 463-447.
-
Edwards A, Grover S, Fishman GA. Frecuency of photographically apparent optic disc and parapapillary nerve fiber layer drusen in Usher Syndrome Retina 1996; 16: 388-392.
-
Shiono T, Noro M, Tamai M. Presumed drusen of optic nerve head in siblings with Usher syndrome. Jpn J Ophthalmol 1991; 35: 300-305.
-
Mets MB, Young NM, Pass A, Lasky JB. Early diagnosis of Usher Syndrome in children. Trans Am Ophthalmol Soc 2000; 98: 237-42.
-
Jaijo T, Aller E, Beneyto M, Nájera C, Millán JM. Estudio genético molecular del síndrome de Usher en España. Acta Otorrinolaringol Esp 2005; 56: 285-289.
-
Espinós C, Millán JM, Bneyto M, Nájera C. Epidemiology of Usher syndrome in Valencia and Spain. Community Genet 1998; 1: 223-228.
-
Yan D, Liu XZ. Genetics and pathological mechanisms of Usher Syndrome. J Hum Genetics 2010; 55: 327-335.
-
Saihan Z, Webster AR, Luxon L, Bitner-Glinzicz M. Update on Usher Syndrome. Curr Opin Neurol 2009; 22: 19-27.