SEMINARIO DE CASOS CLÍNICOS
Manifestaciones oculares del síndrome de Kabuki
GARCÍA CATALÁN MR1, SANTOS-BUESO E2, GARCÍA-FEIJOÓ J2
Unidad de Neurooftalmología. Hospital
Clínico San Carlos. Madrid. España.
1 Licenciado en Medicina y Cirugía.
2 Doctor en Medicina.
RESUMEN
Introducción: El síndrome de Kabuki (SK) es una enfermedad congénita de causa desconocida que se diagnostica en función de cinco criterios clínicos: facies característica, alteraciones en los dermatoglifos, retraso mental y alteraciones esqueléticas y del crecimiento.
Caso clínico: Presentamos un caso de SK en una paciente de 18 años con alteraciones oculares: cejas altas y arqueadas, aumento de la hendidura palpebral, ptosis del ojo izquierdo, hipertelorismo, telecanto, microcórnea, astigmatismo moderado y aumento de la paquimetría corneal.
Conclusión: Aportamos un caso de SK con aumento de la paquimetría corneal. Esta asociación no había sido recogida en la literatura hasta ahora.
Palabras clave: Anomalías oculares, síndrome, paquimetría, retraso mental, Kabuki.
INTRODUCCIÓN
El síndrome de Kabuki (SK) es una rara enfermedad congénita de etiología desconocida y cuya prevalencia se estima en 1 de cada 32.000 en la población japonesa. Se han descrito menos de 400 casos el mundo, siendo 74 de ellos caucásicos. La proporción varón/mujer es similar (1).
El diagnóstico de este síndrome se basa en cinco características: facies peculiar, alteraciones dermatoglíficas, anomalías esqueléticas, retraso mental y alteración del crecimiento con estatura baja (2) (tabla 1).

Los pacientes presentan una cara similar a la de los actores del teatro Kabuki (teatro tradicional japonés) con una hendidura palpebral amplia, pestañas alargadas, cejas muy arqueadas, puente nasal ancho, orejas prominentes, micrognatia, retrognatia e inserción posterior de la línea del pelo (2,3).
Los hallazgos sistémicos frecuentemente asociados son: hipotonía muscular generalizada, laxitud articular, susceptibilidad a padecer infecciones, deficiencias auditivas, defectos cardíacos congénitos y alteraciones del tracto genitourinario (2).
Se presenta el caso de una paciente española de 18 años que, diagnosticada de SK, presenta alteraciones oftalmológicas.
CASO CLÍNICO
Paciente de 18 años diagnosticada de SK derivada a la Unidad de Neurooftalmología para control y seguimiento.
Las características sistémicas eran las siguientes: retraso psicomotor manifiesto, retraso importante del crecimiento, dermatitis atópica, metrorragia funcional, gastritis y duodenitis crónica, pseudo-pseudohipoparatiroidismo, púrpura trombopénica idiopática y anemia ferropénica, así como la facies característica del síndrome de Kabuki: pabellones auriculares grandes y despegados, implantación posterior y triple del cabello, hendiduras palpebrales anguladas, cejas arqueadas, raíz nasal prominente y labio superior fino (fig. 1).
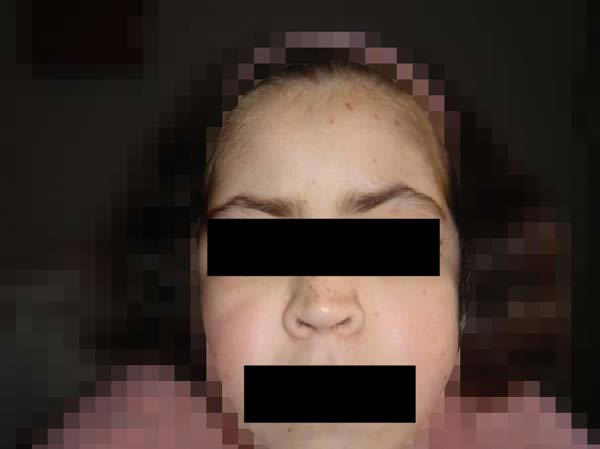
Fig. 1: Facies característica del síndrome de Kabuki.
La hendidura palpebral era alargada presentando hipertelorismo, telecanto y una marcada ptosis del OI.
La agudeza visual sin corrección era de 0.1 en ambos ojos que con corrección (ojo derecho (OD): + 0.5 -4.00 a 90º y ojo izquierdo (OI): +1.5 -4.00 a 70º) mejoraba a 0.6 en ambos ojos.
En la topografía se ponía de manifiesto un astigmatismo irregular. La córnea era irregular con aplanamiento en la zona temporal inferior, con un diámetro de 9 mm (microcórnea) y un grosor de 639 micras en OD y 615 micras en OI.
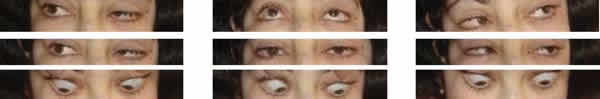
Fig. 2: Motilidad ocular extrínseca normal. Ptosis izquierda.
DISCUSIÓN
En la actualidad no se ha determinado ninguna causa molecular concreta responsable del SK. La aparición esporádica del síndrome sugiere una mutación de novo autosómica dominante. Se han identificado alteraciones cromosómicas como son: duplicación de 1p13.1p22.1, inversión en el brazo corto del cromosoma 4, monosomía parcial 6q, trisomía parcial 12q, translocaciones entre 15q y 17q, delección de novo en 20p12.1 (mutación en el gen C20orf133) y duplicación en la región 8p22-8p23.1 (1).
Las manifestaciones oculares sugestivas del síndrome son: cejas muy arqueadas, epicanto, hipertelorismo, aumento del diámetro horizontal de la hendidura palpebral (más significativo en edades jóvenes), eversión del párpado inferior en el tercio lateral y lagoftalmos. El lagoftalmos nocturno ocurre en más de la mitad de los pacientes con Kabuki y puede predisponer al desarrollo de queratitis o conjuntivitis (3). En nuestro caso existía epicanto acompañado de un aumento de la hendidura palpebral (tabla 2).

Existe un gran número de síndromes congénitos asociados con acortamiento de la fisura palpebral (síndrome alcohólico fetal, síndrome de Dubowith, síndrome de William, anquilobleron y blefarofimosis). Sin embargo, una hendidura palpebral alargada es un rasgo característico del SK (5).
Sakurai describió un caso de Kabuki en el que realizó una reparación quirúrgica del aumento de la hendidura palpebral, mediante la corrección simultánea del canto medial y lateral. En la RMN preoperatoria no encontró alteraciones orbitarias ni del ligamento cantal lateral. Durante la cirugía, observó la existencia de displasia del rafe preseptal orbicular, por lo que optó por la coaptación de la parte lateral del músculo orbicular preseptal, tarsorrafa lateral y Z-plastia lateral y del canto medial, con un buen resultado postoperatorio (4).
Otras manifestaciones oculares son: ptosis, como la que presentaba nuestra paciente, estrabismo (se ha descrito un déficit en la abducción bilateral, endotropía e hipertropía), escleras azules, miopía, ambliopía, Marcus Gunn, nistagmus, microftalmia, opacidad corneal, colobomas iridianos, retinianos y del nervio óptico, papilas oblicuas y pálidas (2,3,5). También se ha descrito la existencia de un caso de SK con telangiectasias retinianas similares a las que aparecen en la enfermedad de Coats (5).
Kluijt hizo una revisión de las manifestaciones oculares en 200 pacientes diagnosticados de SK. Las características más frecuentes eran el estrabismo y las escleras azules seguido de la ptosis y de las alteraciones en la refracción (2).
En conclusión, el SK se diagnostica en función de las características clínicas. Los oftalmólogos deben conocer este síndrome para detectar las alteraciones oculares asociadas y tratarlas precozmente para evitar el deterioro visual de los pacientes.
BIBLIOGRAFÍA
-
Maas NMC, Van de Putte T, Melotte C, Francis A, Schrander-Stumpel C, Sanlaville D et al. The C20orf133 gene is disrupted in a patient with Kabuki síndrome. J Med Genet 2007; 44: 562-569.
-
Kluijt I, van Dorp DB, Kwee MLK, Toutain A, Keppler-Noreuil K, Warburg M et al. Kabuki syndrome. Report of six cases and review of the literature with emphasis on ocular features. Ophthalmic Genet 2000; 21: 51-61.
-
Chaudhry IA, Shamsi F, Alkuraya HS, Al-Sharif A. Ocular manifestations in Kabuki syndrome: the first report from Saudi Arabia. Int Ophthalmol 2008; 28: 131-134.
-
Sakurai H, Nozaki M, Takeuchi M, Soejima M, , Kajimoto M, Hori S. Periorbital correction in Kabuki syndrome. Plastic and reconstructive surgery 2003; 111: 1461-1464.
-
Anandan M, Porter NJ, Nemeth AH, Blair E, Downes S. Coats-type retinal telangiectasia in case of Kabuki make-up syndrome (Niikawa-Kuroki syndrome). Ophthalmic Genet 2005; 26: 181-183.