SEMINARIO DE CASOS CLÍNICOS
Síndrome de Axenfeld-Rieger. ¿Glaucoma o anomalía de los discos ópticos?
PERUCHO MARTÍNEZ S1, FERNÁNDEZ ESCÁMEZ CS2, MARTÍN GIRAL E3
Hospital Universitario de
Fuenlabrada. Servicio de Oftalmología. Fuenlabrada, Madrid.
1 Doctora en Medicina y Cirugía.
2 Licenciado en Medicina y Cirugía.
3 Licenciada en Medicina y Cirugía.
RESUMEN
Introducción: El Síndrome de Axenfel-Rieger es una rara enfermedad congénita del desarrollo ocular que clásicamente se traduce en una disgenesia del segmento anterior.
Sin embargo, al tratarse de una anomalía en el desarrollo no debemos olvidar que podemos encontrar otras alteraciones oculares.
Caso clínico: Paciente varón de 8 años de edad remitido para estudio desde el Servicio de Neurología con diagnóstico de Síndrome de Rieger. Oftalmológicamente el paciente presenta corectopia, policoria, hipoplasia iridiana, embriotoxon posterior en ambos ojos. Agudeza Visual (AV) OD 0,3 y en OI 0,7. Cristalino transparente. Presión intraocular (PIO) OD 24 y PIO OI 20 mmHg. En la resonacia magnética (RMN) de cerebro se observa una asimetría clara de los nervios ópticos en su recorrido orbitario. El nervio óptico derecho es más elongado con una posible ectasia de su vaina y el nervio izquierdo podría ser ligeramente hipoplásico. En su porción intracraneal dichos nervios y el quiasma presentan únicamente asimetría en la disposición sin alteraciones en el calibre o señal.
Palabras clave: Síndrome de Axenfeld-Rieger, glaucoma infantil, neuropatías congénitas.
INTRODUCCIÓN
La presencia de embriotoxon posterior en asociación con procesos iridianos prominentes es conocida como anomalía de Axenfeld. Si se añade una hipoplasia iridiana la condición toma el nombre de anomalía de Rieger. Si a las anomalías oftalmológicas se le suman anomalías y defectos sistémicos entonces estamos ante un Síndrome de Axenfeld-Rieger (1,2).
El Síndrome de Axenfeld-Rieger es una rara enfermedad congénita y progresiva del desarrollo ocular que clásicamente se traduce en una disgenesia del segmento anterior (3-5).
Sin embargo, al tratarse de una anomalía en el desarrollo no debemos olvidar que podemos encontrar otras alteraciones oculares como desprendimiento de retina (6), hipermetropía (7) o hipoplasia del nervio óptico. En este caso se observó una asimetría clara en los nervios ópticos con una elongación el nervio óptico derecho y una posible ectasia de su vaina; y el nervio izquierdo podría ser ligeramente hipoplásico.
CASO CLÍNICO
Paciente varón de 7 años de edad remitido para estudio oftalmológico desde el Servicio de Neurología con diagnóstico de Síndrome de Axenfeld-Rieger.
Desde el punto de vista sistémico el paciente presentaba cardiopatía congénita (comunicación auricular), leve retraso en la maduración del lenguaje, hidronefrosis grado II y anomalía del desarrollo venoso cerebral derecho.
Oftalmológicamente el paciente presenta corectopia, policoria, hipoplasia iridiana, embriotoxon posterior en ambos ojos (fig. 1). AV OD 0,3 y en OI 0,7. Cristalino transparente. PIO OD 24 y OI 22 mmHg. En el fondo de ojo se observó una asimetría de los discos ópticos con cierta palidez del disco óptico izquierdo (fig. 2). La excavación papilar del disco derecho era de 0,1 mientras que la del disco izquierdo era de 0,3. Se realizó perimetría 24-2 no observándose escotomas específicos de glaucoma.
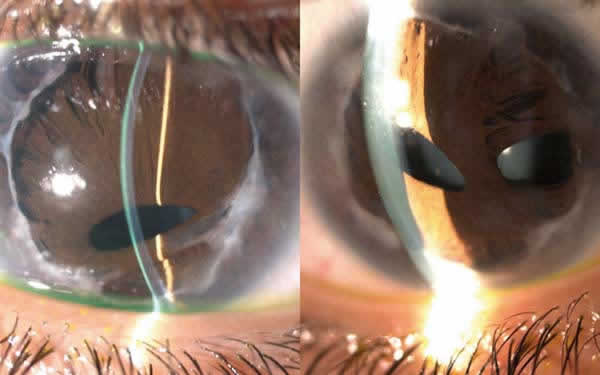
Fig. 1: Embriotoxon posterior OD y OI respectivamente. Policoria, corectopia e
hipoplasia de iris. Cristalino trnasparente.
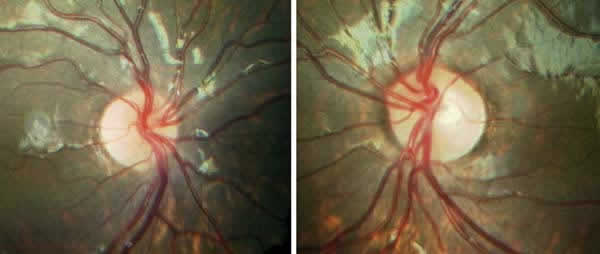
Fig. 2: Papilas de OD y OI respectivamente. Obsérvese cierta palided del disco
óptico izquierdo.
La RMN de cerebro con cortes orbitarios mostró una elongación el nervio óptico derecho y con una posible ectasia de su vaina; y el nervio izquierdo podría ser ligeramente hipoplásico (fig. 3). En su porción intracraneal dichos nervios y el quiasma presentan únicamente asimetría en la disposición sin alteraciones en el calibre o señal.
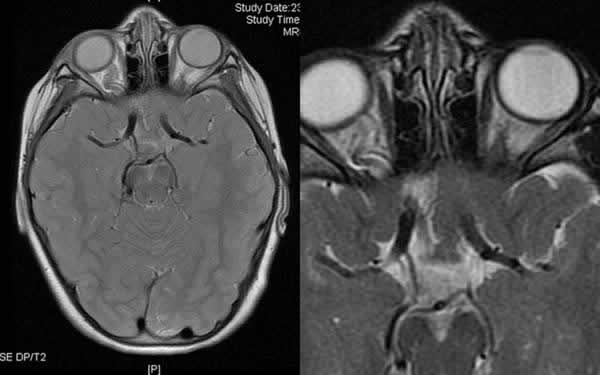
Fig. 3: RMN cerebral. Asimetría de ambos nervios ópticos, claramente mayor el
derecho (posible elongación de la vaina del derecho frente a posible atrofia del
izquierdo).
Actualmente el paciente tiene 9 años y la AV con su corrección (OD +6,00 – 2,75 x 170° y OI +4,00 – 1,75 x 180°) es de 0,8 en OD y de 1 en OI. La PIO en OD es 17 y en OI 16; no tiene tratamiento hipotensor y es sometido a revisiones periódicas en consulta de Oftalmología y de Neurología para seguimiento de la anomalía de los discos ópticos y la detección de complicaciones asociadas.
DISCUSIÓN
En nuestro caso se trataba de un Síndrome de Axenfeld-Rieger. Otras anomalías que pueden encontrarse son: estrabismo, catarata, quistes dermoides, desprendimiento de retina, degeneración macular, colobomas coriorretinianos y muy raramente hipoplasia coroidea (1-6).
El glaucoma se desarrolla hasta en un 50% de los casos y frecuentemente durante la infancia media-tardía (8). El glaucoma se produce por la asociación de la anomalía angular con un cierre angular secundario a sinequias.
Todos estos hallazgos oculares están ampliamente descritos en la literatura y se sabe que la condición, puede conducir a una ceguera gradual e irreversible, especialmente por el glaucoma asociado de muy difícil manejo (9-11).
Sin embargo no debemos olvidar que ante todo, es un fallo disgenésico y una anomalía en el desarrollo ocular. Por ello, debemos siempre buscar otras alteraciones no tan frecuentes como la alteración (hipoplasia, elongación) del nervio óptico, que en algunos casos nos puede hacer pensar en patología glaucomatosa sin que ésta esté presente, al menos por el momento.
En estos pacientes se recomienda hacer un estudio completo que incluya técnicas de neuroimagen que nos ayuden a hacer un diagnóstico correcto de las anomalías oculares.
Por otra parte, se trata de pacientes que deben ser seguidos muy de cerca para detectar cualquier cambio o desarrollo de complicaciones. En este caso clínico, el diagnóstico de glaucoma todavía no está establecido pero podría estar entorpecido por presentar unos discos ópticos ya alterados en los cuales no podríamos apoyarnos para el seguimiento.
CONCLUSIONES
Se trata de una enfermedad poco frecuente pero hasta con un 50% de asociación glaucomatosa (8). No debemos olvidar que al ser un síndrome malformativo otras entidades pueden asociarse y hacernos malinterpretar algunos signos clínicos como en este caso la asimetría del disco óptico e hipertensión ocular. Las pruebas de neuroimagen en estos casos son imprescindibles.
En este caso hemos optado por la vigilancia y seguimiento mediante PIO, tomografía de coherencia óptica (OCT) de fibras nerviosas, tomografía retiniana de Heidelberg (HRT II) y perimetría.
Si aparece glaucoma el tratamiento inicialmente será medicamentoso, aunque en algunos casos habrá que acudir a la goniotomía o cirugía filtrante dependiendo de cada paciente.
BIBLIOGRAFÍA
-
Koshino T, Konno T, Ohzeki T. Bone and joint manifestations of Rieger's syndrome: a report of a family. J Pediatr Orthop 1989; 9(2): 224-30.
-
Cunningham ET Jr, Eliott D, Miller NR et al. Familial Axenfeld-Rieger anomaly, atrial septal defect, and sensorineural hearing loss: a possible new genetic syndrome. Arch Ophthalmol 1998; 116(1): 78-82.
-
Goddé-Jolly D, Dufier JL. Oftalmología pediátrica. Barcelona: Masson; 1994; 154-64.
-
Shields MB. Atlas de glaucoma. Buenos Aires: Waverly Hispánica; 1998; 1: 70-5.
-
Kulak SC, Kozlowski K, Semina EV et al. Mutation in the RIEG1 gene in patients with iridogoniodysgenesis syndrome. Hum Mol Genet 1998, 7(7): 1113-7.
-
Spallone A. Retinal detachment in Axenfeld-Rieger syndrome. Br J Ophthalmol 1989; 73(7): 559-62.
-
Spierer A, Barak A. Anterior segment pathology associated with hypermetropia. Ophthalmic Genet 1996; 17(2): 79-83.
-
Dhir L, Frimpong-Ansah K, Habib NE. Missed case of Axenfeld-Rieger syndrome: a case report. Cases J. 2008 Nov 6; 1(1): 299
-
Honkanen R, Alward WL. Cossari AJ. Progressive iris changes in a case of Axenfeld-Rieger syndrome. Arch Ophthalmol 2006. Dec; 124(12): 1793.
-
Tasman W, Jaeger EA, eds. Duane’s Clinical Ophthalmology. Philadelphia: Lippincott; 2002.
-
Walton DS, Katavounidou G. Newborn primary congenital glaucoma: 2005 update. J Pediatr Ophthalmol Strabismus 2005; 42(6): 333.341.