SEMINARIO DE CASOS CLÍNICOS
Epiteliopatía pigmentaria Placoide. Seguimiento evolutivo y controversia ante su tratamiento
ECHEVARRÍA LUCAS L1, NIEVAS GÓMEZ T1
1
Doctora en Oftalmología por la Universidad de Málaga. Servicio de Oftalmología
del Hospital de la Axarquía (Vélez-Málaga). Servicio Andaluz de Salud.
2 Jefa de Servicio de Oftalmología del Hospital de la Axarquía
(Vélez-Málaga). Servicio Andaluz de Salud.
RESUMEN
Introducción: La epiteliopatía pigmentaria placoide (EPPMPA) es una patología infrecuente idiopática bilateral que afecta a individuos entre 20 y 40 años. Puede iniciarse tras enfermedad viral, desarrollando vasculitis coroidea con reacción secundaria en el epitelio pigmentario retiniano; es normalmente autolimitada, aunque a veces puede producir pérdida visual y, sobre todo, vasculitis cerebral grave. Se discute el uso profiláctico de corticoides orales.
Caso clínico: Paciente de 40 años que acude a urgencias con cuadro de fotopsias en ambos ojos tras episodio gripal. No presentaba pérdida visual, pero la funduscopia mostraba múltiples lesiones placoides de color gris crema o blanco, localizadas alrededor de los vasos. Ante cuadro tan florido se plantea el uso de corticoides orales de entrada, resolviéndose el proceso sin cicatrices epiteliales y sin afectación en el sistema nervioso central.
Palabras clave: Epiteliopatía pigmentaria placoide (EPPMPA), vasculitis cerebral y coroidea, corticoides orales, ciclosporina.
INTRODUCCIÓN
La epiteliopatía pigmentaria placoide (EPPMPA) es una patología inflamatoria infrecuente idiopática bilateral que afecta a individuos de ambos sexos por igual entre 20 y 40 años (1,2). En su patogenia destaca que los antígenos HLAB27 y HLADR2 (3) son más frecuentes en pacientes que desarrollan esta respuesta inflamatoria. Entre 25 y 40% de los casos (4,5) presentan pródromo de enfermedad viral, tras la cual se desarrolla una vasculitis coroidea con una reacción secundaria en el epitelio pigmentario retiniano (5). Se han descrito, junto con la EPPMPA otras patologías sistémicas como tuberculosis, parotiditis, granulomatosis de Wegener, poliarteritis nodosa, colitis ulcerosa, sarcoidosis y enfermedad de Lyme (6), así como antecedentes de vacunación anti-hepatitis B.
La EPPMPA se presenta con fotopsias, escotomas y pérdida de visión subaguda (6). Ambos ojos pueden ser afectados pero de manera asimétrica. La pérdida visual depende de la presencia de lesiones bajo la fóvea. La visión puede ser desde normal a 20/200 o peor. Hay escotomas del campo visual que corresponden con lesiones del fondo. Puede haber defectos pupilares aferentes pero de manera asimétrica (1,5). En la exploración del polo posterior se hallan lesiones crema o gris blanco a nivel del epitelio pigmentario retiniano que empiezan en polo posterior y se extienden al fondo del ojo postecuatorial. En unos días, las lesiones van desapareciendo a nivel central, pero sin mejoría inmediata de la visión. En dos semanas las lesiones agudas son sustituidas por cambios en el EPR de diversa intensidad. Pese a todo pueden aparecer nuevas lesiones en distintos estadios de evolución. En unos meses la agudeza visual se recupera pero a veces los escotomas paracentrales pueden persistir (5,6).
La vasculitis cerebral (5) se asocia con la EPPMPA. Pueden tener leucocitosis cerebral junto con dolores de cabeza y otros hallazgos en el sistema nervioso central. Por esta asociación, debe realizarse una RNM en pacientes con EPPMPA. El tiempo entre la aparición de EPPMPA y la vasculitis cerebral es de unos 3 meses. La AFG muestra hipofluorescencia precoz con ausencia de perfusión coriocapilar e hiperfluorescencia tardía (7).
Mediante la técnica de verde de indocianina se halla una vasculitis oclusiva, que en casos severos afecta al EPR y otras partes de la retina. Otros hallazgos son la alteración del ERG y del EOG, ambos reducidos, prolongándose la adaptación a la oscuridad. Estas alteraciones suelen resolverse en un año (5). Entre las secuelas de EPPMPA puede darse rara vez neovascularización. Si los cambios se producen bajo la fóvea, puede limitarse la visión.
El tratamiento sólo está indicado en caso de vasculitis cerebral o visión marcadamente disminuida. En estos casos los corticoides pueden ser efectivos. En caso de falta de respuesta a los corticoides, se podría administrar ciclosporina (6,8,9).
CASO CLÍNICO
Una paciente de 40 años acude a urgencias por presencia de fotopsias en ambos ojos. Comenta haber sufrido un episodio gripal en las últimas dos semanas. No refiere cefaleas ni síntomas neurológicos. Presenta leves defectos pupilares aferentes. La AV era 1 en ambos ojos. La biomicroscopía no mostraba alteraciones de interés y el tyndall era negativo. En la funduscopia de ambos ojos destaca la presencia de lesiones placoides múltiples de color gris crema o blanco, localizándose alrededor de los vasos y sobre ellos en las arcadas superiores e inferiores y rodeando los mismos, siguiendo su trayecto hasta la periferia. En polo posterior se observa cerca alguna lesión, pero siempre a más de 500 μ de la mácula (figs. 1a y b).
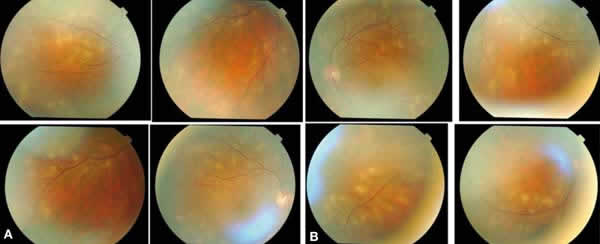
Fig. 1: En ambos ojos destacan
lesiones placoides múltiples de color gris crema o blanco, alrededor de los
vasos y sobre ellos en las arcadas superiores e inferiores y rodeándolos,
siguiendo su trayecto hasta la periferia.
Tras descartar patología infecciosa (tuberculosis, Lyme, sífilis…) mediante una batería diagnóstica, la paciente fue tratada con 60 mg. de prednisona y citada posteriormente para realizarle fotos de control (figs. 2a y 2b) y una AFG (fig. 3), observándose una leve mejoría de las lesiones, que aparecen más difuminadas y separándose de los vasos. El polo posterior permanece limpio. Una semana después se cita nuevamente a la paciente, observándose la reabsorción de las lesiones, quedando aún algunas activas pero sin cicatriz en el EPR. A la vez, aparecen otras nuevas (fig. 4). El corticoide, no obstante, se va retirando a razón de 5 mg. por semana hasta suspenderlo en el plazo de un mes. Se solicita RNM y exploración neurológica, así como una batería diagnóstica complementaria completa para descartar otras posibles patologías: sarcoidosis, oftalmía simpática, infiltrados coroideos metastásicos… Un mes después se observan sólo algunos rastros de las lesiones, que desaparecen sin dejar pigmentación en el EPR (fig. 5). Y pasadas tres semanas más se observa sólo algún leve rastro (fig. 6). El resultado de las pruebas complementarias fue normal, incluido el de la RNM, que no muestra signos de vasculitis cerebral.

Fig. 2: A la semana siguiente, se
observa una leve mejoría de las lesiones, que aparecen más difuminadas y
separándose de los vasos.
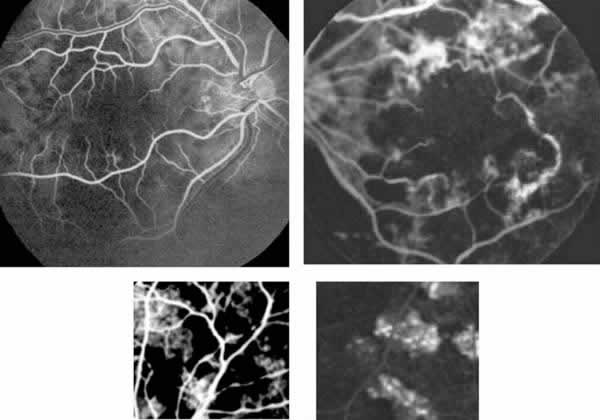
Fig. 3: AFGs: Arriba, izquierda,
hipofluorescencia típica de lesiones en tiempos precoces (OD). Arriba, derecha,
defectos del epitelio pigmentario y vasculitis OI sin edema central. Abajo,
detalles de las placas inflamatorias con hipofluorescencia precoz e
hiperfluorescencia tardía.
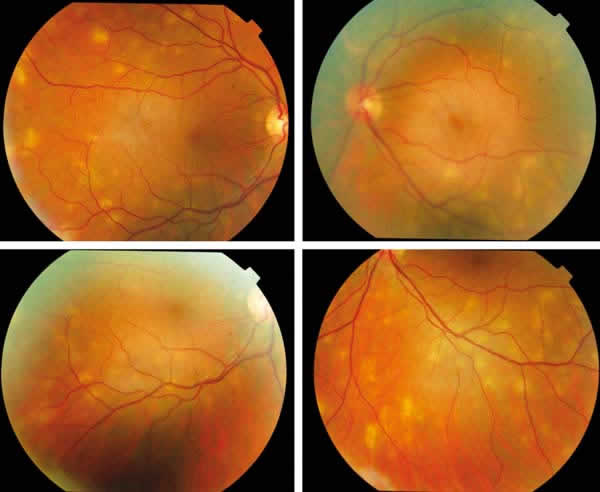
Fig. 4: Una semana después, se cita
nuevamente a la paciente, observándose la reabsorción de las lesiones, quedando
aún algunas activas pero sin cicatriz en el EPR. A la vez, aparecen otras
nuevas.
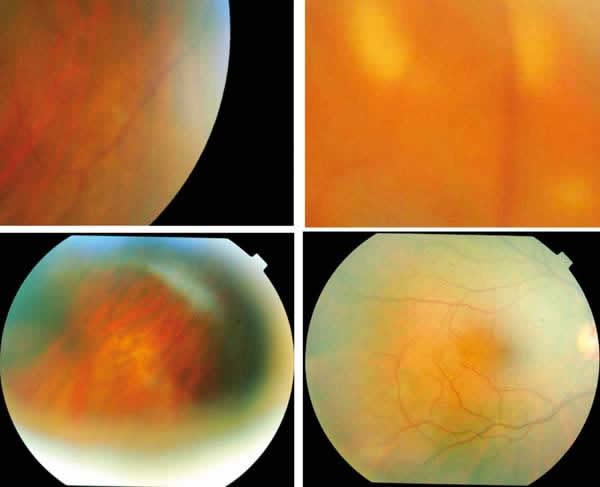
Fig. 5: Un mes después se observan
sólo algunos rastros de las lesiones, que desaparecen sin dejar pigmentación en
el EPR.
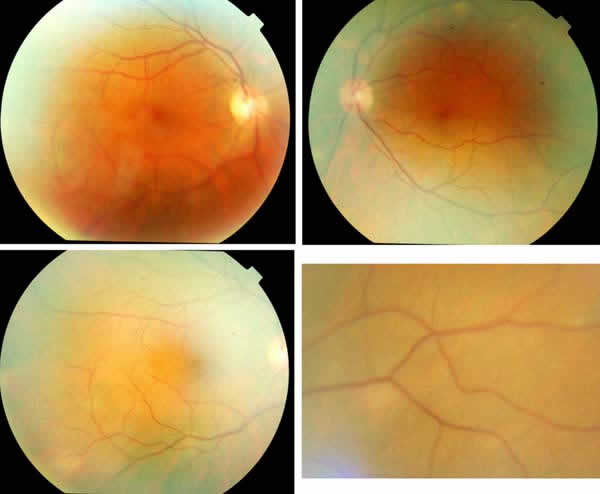
Fig. 6: Pasadas tres semanas más se
observa sólo algún leve rastro.
DISCUSIÓN
La EPPMPA es una enfermedad en principio autolimitada (1), aunque es importante un correcto diagnóstico diferencial (5) con otras enfermedades de aspecto similar pero mucho peor pronóstico, en que la introducción precoz de corticoides o inmunosupresores podría frenar la pérdida visual: coroidopatía serpiginosa, coroiditis multifocal con panuveítis, enfermedad de Harada, oftalmía simpática,… En otras enfermedades de aspecto similar, como sarcoidosis, sífilis secundaria o infiltrados coroideos metastásicos es importante su filiación, puesto que su desconocimiento puede costar la vida. Otra patología que forma parte del diagnóstico diferencial de la EPPMPA es el síndrome de múltiples puntos blancos evanescentes que tiene un curso benigno similar.
Factores de riesgo que se consideran de mal pronóstico visual en su evolución son: implicación foveal al comienzo de la patología, edad avanzada al comienzo, unilateralidad, bilateralización tras más de seis meses, recurrencia, y filtración de venas coroidales (6).
Es controvertido el uso de corticoides (5,6,8,9) en la EPPMPA desde el inicio de su diagnóstico; en principio estaría indicado sólo en caso de vasculitis cerebral o pérdida visual severa. Pero cada vez son más los expertos que recomiendan su introducción profiláctica para evitar la aparición de vasculitis cerebral (5,6,8) o para resolver más rápidamente la inflamación, mejorando el pronóstico visual final. Si no hay respuesta a los corticoides, se podría administrar ciclosporina (5,9).
CONCLUSIONES
La EPPMPA es una enfermedad en principio autolimitada y de buen pronóstico visual. Es una vasculitis coroidea oclusiva que, en casos más severos, puede afectar al EPR o a otras partes de la retina. Entre un 25-40% de los casos es una respuesta secundaria a un proceso viral. Pero no es un cuadro que se deba menospreciar, pues en un pequeño porcentaje de casos puede desarrollar una vasculitis cerebral que debe ser correctamente diagnosticada y tratada.
El caso que nos ocupa tuvo muy buena evolución y no quedaron restos de alteraciones pigmentarias residuales. En esta paciente se introdujo una tanda de corticoides desde el principio, que podría (o no) tener relación con la buena evolución final y la no aparición de lesiones en polo posterior. El SNC no fue afectado, presentándose una RNM sin alteraciones. Es necesario un correcto diagnóstico diferencial con enfermedades de aspecto similar pero mucho más graves, que deben de identificarse y tratarse precozmente.