CAPÍTULO 4 SÍNDROME DE SJÖGREN PRIMARIO Juan Murube |
| DEFINICIÓN El síndrome de Sjögren* primario (SS1) es una enfermedad sistémica caracterizada por destrucción autoinmune de las glándulas exocrinas, que produce un deficit secretorio externo que puede afectar a todo el cuerpo. En él suelen encontrarse anticuerpos contra las ribonucleoproteínas SSA y SSB y la a -fodrina, e infiltración linfocitaria de las glándulas.
HISTORIA El síndrome de Sjögren, si por ello entendemos lo que los autores que hace unos 60 años le dieron tal nombre, -es decir, una sequedad ocular crónica asociada a otras sequedades exocrinas; más tarde se incluyó la artritis reumatoide que no empezó a ser identificado hasta finales del siglo pasado. En la literatura de la antigüedad grecolatina se cita varias veces la asociación de xeroftalmía y reuma (Galeno s II AD a; Pablo de Egina s VII AD) lo que ha hecho pensar que ya se conocía la asociación sindrómica de ojo seco y artritis reumatoide sin embargo, en griego, reuma era una secreción patológica, fuese mucosa, purulenta o de otro tipo, y no una enfermedad articular. Así, cuando Pablo de Egina dice que la xeroftalmía es una afección pruriginosa sin reuma, quiere decir sin descarga ocular inflamatoria. En el siglo pasado, Fischer 1883 publicó un caso de querotopatía filamentosa bilateral asociada a artritis deformante; Fraser 1893, el de una mujer de 27 años que llevaba 3 años y medio padeciendo intensa sequedad de ojos, nariz y boca y Mikulicz 1892 un cuadro de hinchazón simétrica y simultánea de las glándulas lacrimales y salivales. Gougerot 1925 publicó la asociación sindrómica de sequedad ocular, bucal y vaginal. Houwer 1927 comunicó 9 casos de queratitis filamentosa bilateral asociada a artritis crónica de los 9 casos, 7 se daban en mujeres adultas, la mayoría de ellas menopáusicas; en la discusión; Clegg refirió un caso similar de una mujer con oftalmoxerosis y reumatismo. Isakowitz 1928 reportó la queratitis filiforme por hiposecreción lacrimal asociada a artritis de ambas manos y pies, en una mujer menopáusica. Henrik Sjögren (1930) comunicó la asociación sindrómica de sequedad ocular y bucal, y más tarde (1933), su posible asociación con artritis. Este síndrome de exocrinopatía múltiple, asociado o no a artritis reumatoide, comenzó al poco a ser llamado síndrome de Sjögren (von Grosz 1936, Hamilton 1940, de Roetth 1945), de Gougerot-Sjögren (Wechsler et al 1976, Liotet et al 1987), de Gougerot-Houwers-Sjögren (Paganoni 1961) o de Mikulicz-Sjögren (Bain 1960). El nombre de síndrome de Sjögren se aplica también a la asociación sindrómica de catarata congénita y oligofrenia (T.Sjögren 1935). Holm 1949 prefería hablar de syndrome sicca. Pero el término de síndrome de Sjögren para referirse a la exocrinoxerosis, asociada o no cualquier tipo de reumatismo articular, prevaleció y extendió grandemente (Talal et al 1975, Strom et al 1978, Moutsopoulos et al 1978, 197), hasta el extremo de ser aplicado por los años 40 y 50 a todas las KCS de causa desconocida estuviesen o no asociadas a otras exocrinoxerosis. Más tarde se descubrió que las glándulas exocrinas de los pacientes del síndrome están infiltradas por numerosos linfocitos. En 1951, Rothman et al publicaron que el síndrome se complica frecuentemente con linfoblastomas malignos. Escande 1977 propuso introducir el término de syndrome lymphoexocrinus para la sequedad ocular asociada sindrómicamente a conectivopatías, pues todas ellas tienen el factor común de invasión linfocitaria y Talal 1984, el de exocrinopathia autoimmune, para expresar que es una infiltración linfoidea de las glándulas de secreción externa. Al mismo tiempo se fue descubriendo que la asociación a artritis reumatoide puede estar substituida por asociaciones a otras conectivopatías autoinmunes, como lupus eritematoso sistémico, esclerodermia y polimiositis. En la década de los 70 se descubrió la frecuente asociación del SS a ciertos antígenos HLA (Fye et al 1976, Hinzowa et al 1977, Chused et al 1977, etc) y la coexistencia con determinados autoanticuerpos (Anderson et al 1961, Alspaugh et al 1976, Akizuki et al 1977, Kassan et al 1977). Al observarse que el substrato antigénico del síndrome era distinto en los pacientes con y sin conectivopatía sistémica, Frost Larsen (1978) propuso denominar SS primario al que a las manifestaciones exocrinas típicas no añade una artritis reumatoide u otras conectivopatías autoinmunes, y SS secundario, al que sí las añade. Aunque esta denominación es muy inapropiada (pues puede inducir a pensar que el SS secundario es secundario al SS primario, lo que no es cierto), lo cierto es que se extendió grandemente y es la que se usa actualmente. Sería preferible llarmalos SS1 y SS2.
EPIDEMIOLOGÍA El SS afecta a todas las razas, climas y países. El 90% de los casos aparece en mujeres menopáusicas; el otro 10% en hombres o en jóvenes de uno u otro sexo. Balsa (1996) expone que los dos principales inconvenientes para conocer bien la frecuencia del síndrome de Sjögren son : (1) La aplicación de distintos criterios diagnósticos. Una persona que viva en Dinamarca pueder ser dignósticada de SSciertos criterios diagnósticos; sin embargo, otra persona con idénticos síntomas y viviendo en EEUU no sería diagnosticada de Sjögren al aplicarse criterios distintos. (2) Los pacientes con SS pueden ser vistos por diversos especialista (reumatólogos, oftalmólogos, médicos de familia, internistas), por lo que es difícil conocer el número de pacientes nuevos que ve cada uno de ellos y el número de pacientes que doblan con otros especialistas. En España, Riaza 1960 calculó que el SS afecta al 0'53 por mil de los enfermos oftalmológicos, cifra hoy considerada muy baja, y propia de una época en que la mayoría de los casos no se diagnosticaban. Actualmente, en España, con una población de 40 millones de habitantes, hay aproximadamente 400.000 síndromes de Sjögren, la mayoría de ellos con formas ligeras indiagnosticadas; de ellos, aproximadamente la mitad padecen SS1 y la otra mitad SS2. Casi todos los SS2 van asociados a artritis reumatoide. Desde el punto de vista de la especialidad reumatológica, en España hay aproximadamente 1 millón de pacientes de artritis reumatoide; de ellos, el 20%, es decir, 200.000, conllevan una xerosis exocrina. Para Balsa 1996, el 30% de los pacientes con artritis reumatoide padecen un SS. Con ser el SS bastante frecuente y muy conocido, no es el más frecuente de los cuadros clínicos de sequedad ocular (Illa et al, 1993).
ETIOPATOGENIA Teoría de la denervación parasimpática. La causa del SS se pensó hace años que sería un trastorno neurológico que provocaría una denervación parasimpática paulatina de las glándulas (Holm 1949, Sautter 1949, Weber 1957). Esta teoría fue atacada por De Haas 1961a, quien observó que que los enfermos de SS no responden a la administración subcutánea de simpaticomiméticos como ocurriría en una glándula lacrimal denervada por sección facial. Teoría endocrina. Se apoya en el hecho clínico de la frecuente aparición de la enfermedad en mujeres, especialmente en menopáusicas y ovariectomizadas. Inicalmente se pensó que la causa sería un defecto de estrógenos, y más tarde se pasó a pensar que esto era accidental, y que el déficit hormonal causal es el defecto de andrógenos que acompaña al climaterio. Se piensa hoy que el papel de los factores hormonales no es fundamental, sino modulador. Teoría carencial. Se apoyó en las coincidencias clínicas del SS con el síndrome de Plummer-Vinson. Uno de los factores etiológicos de este último es la carencia de vitamina B12. También se han encontrado ciertas analogías clínicas entre el SS y la arriboflavinosis, a lo que se opuso Lutman et al 1946, pues la arriboflavinosis cura con la administración de vitamina B2 o de complejo vitamínico B, mientras que el SS no. Teoría genética. En el SS hay a veces una evidente transmisión genética. Así, Lisch 1937 publicó un genodendro con 12 casos en 3 generaciones, Coverdale 1948 un genodendro con un padre y su hija y otro con una madre y su hija, François 1958 un genodendro con una madre y su hija, etc. Sin embargo nunca se ha descrito un SS bien definido en dos hermanos gemelos homozigotes. La frecuente incidencia de SS1 en sujetos con aloantígenos de histocompatibilidad HLA B8 y DR3 y del SS2 en sujetos con DR4 refuerza la presunción del factor genético. Se están descubriendo características genéticas propias de algunos SS, como que la matriz extracelular de los lacrimocitos aparece alterada por modificaciones del gen BM180 y de la gliadina, que rigen la formación de la laminina-1 (Asrani et al 1996), o que la proteolisis de la a -fodrina en los SS tiene una base genética (Haneji et al 1977). Teoría infecciosa. La existencia
de factores infecciosos se sospecha al menos en el SS1, por su frecuente asociación con
la presencia en el suero del paciente del Ac anti-SS-B. La ribonucleoproteína SS-B se
encuentra en el genoma del virus de Epstein-Barr, y se piensa que estos genes virales
pueden activar los linfocitos B de los sujetos infectados y ser la causa de la
linfoproliferación que presentan estos pacientes (Lerner et al 1981). Los pacientes de SS tienen una hiperreactividad delos linfocitos B, que se transforman en plasmocélulas, las cuales, además de formar inmunoglobulinas contra los antígenos de los epitelios exocrinos, las formarían contra los linfocitos T-supresores. La depresión de los linfocitos T-supresores perpetuaría la hiperreactividad de los linfocitos B y la agresión autoinmune. En apoyo de esta suposición está la comprobación de la hiperreactividad de los linfocitos B (hipergammaglobulinemia, macroglobulinemia de Waldenström), la depresión de los linfocitos T-supresores, y la activación de los linfocitos T-helper. Gratwohl et al 1977 observaron en pacientes a los que se había hecho un transplante de médula ósea, con reacción crónica transplante-versus-huésped, que desarrollaban un año después un SS (xeroftalmía, xerostomía, infiltrados linfocitarios en glándulas labiales) sin tener antígenos HLA-B8 ni anticuepos anti-ENA. Se supone que la reacción transplante-versus-huésped activa linfocitos que, por una razón desconocida, destruyen lás glándulas exocrinas. Esto ha llevado a pensar que en el SS2 puede ocurrir algo parecido, con la salvedad de que la reacción la provoca la artritis reumatoide, la cual suele preceder años o meses al deterioro y destrucción exocrinos. Los pacientes de SS presentan hipergammaglobulinemia producida por aumento global de todas la inmunoglobulinas mayoritaria, IgG, IgA e IgM (Cave 1979, Dunne et al 1979), con presencia de anticuerpos antinúcleo, antimitocondria, y sobre todo antiepitelio de los canales excretores salivales y lacrimales. La existencia de anticuerpos anti-epitelio de los conductos excretores de las glándulas lacrimales y salivales han sido estudiados por numerosos autores (Anderson et al 1973, Söborg et al 1978, etc). Ichikawa et al 1980 sugirieron que el antígeno desencadenante de la respuesta inmunológica podría ser la pieza secretoria de la IgA secretoria. Zittoun et al 1978 determinaron que estos anticuerpos son una IgG asociada en el 40% de los casos a una IgM, y en el 10% a una IgA. Feltkamp et al 1968, quienes estudiaron lo anticuerpos contra los canales de las glándulas salivales en el SS, vieron que también existen en el 27% de los individuos de edad avanzada. La existencia de anticuerpos antinucleares en el SS fue puesta de manifiesto por Alspaugh et al 1976 y Akizuki et al 1977 Los antigenos HLA A8 , HLA B8, DR3 y DR4 se encuentran con más frecuencia en el SS que en población sana (Gerschwin et al 1975, Chused et al 1977, Powell et al 1980, Moutsopoulos et al 1978b,1980b) El citoesqueleto que queda bajo el plasmalemma de los lacrimocitos contiene abundante cantidad de una proteína, la fodrina. La activación de una proteasa desconocida (como ocurre fisiológicamente en el fenómeno de apoptosis) provoca en el síndrome de Sjögren una protelisis de la fracción a -fodrina, que ya puede detectarse desde los primeros pasos del SS-I, alterando la secreción. La a -fodrina puede ser el autoantígeno organoespecífico que desencadena el SS (Haneji et al 1997). La existencia de lesiones distintas a las de los epitelios exocrinos pueden explicarse de diversas formas. Una de ellas es la de la alteración del receptor del fragmento Fc (fracción cristalizable) de las inmunoglobulinas, en las que es posible que los inmunocomplejos circulantes jueguen un papel importante. Tanto los pacientes de SS1 como de SS2 tienen gran cantidad de inmunocomplejos séricos. Estos inmunocomplejos deberían ser destruidos por los fagocitos mononucleares del sistema reticulo-endotelial que adhieren a su superficie celular los receptores de las Ig. Parece ser que en el SS1, si estos receptores son normales, las manifestaciones del síndrome son sólo glandulares, pero si son anormales, aparecen también manifestaciones extraglandulares, porque los inmunocomplejos circulantes se depositan específicamente en algunos tejidos, provocándoles una reacción inflamatoria; estos inmunocomplejos pueden tener composiciones diversas, y por tanto, ser unos más dañinos que otros. Una situación en cierto modo parecida se ha visto en el lupus eritematoso sistémico, en el que estos receptores son normales cuando la enfermedad está silente, pero anormales, cuando está activa. También se tratan de explicar las lesiones perivasculares de pulmón, piel, nervios, músculos, etc. en ausencia de deposición pasiva de inmunocomplejos. Así, Swartz et al 1986 sugirieron que las células inmunocompetentes de la pared vascular producen inmunoglobulinas, inmunocomplejos o complemento. El complemento podría ser fijado y activado in situ, con la producción de potentes factores quemotácticos que atraerían linfocitos polimorfonucleares e iniciarín una inflamación vascular.
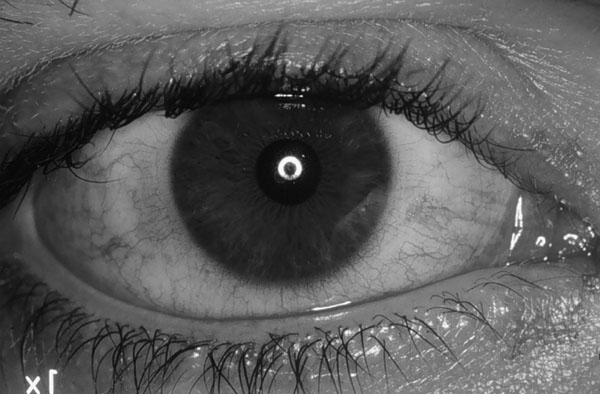
CLÍNICA El SS1 suele aparecer en mujeres de la segunda mitad de la vida, aunque a veces lo hace en varones adultos o en jóvenes hembras o varones. Se inicia de forma subaguda o aguda una xeroftalmía y xerostomía de intensidad ligera, media o grave, que se mantiene estacionaria o avanza de forma recurrente o continua. Ocasionalmente se acompaña de episodios de hinchazón parotídea (paperas). A menudo aparecen otras xerosis exocrinas (xerodermia, aquilia, nariz seca, vagina seca, garganta seca) o manifestaciones de invasiones linfocitarias de otros órganos (sistema retículo-endotelial, vasos sanguíneos, pulmones, riñones, músculos, sistema nervioso). A veces hay episodios de febrícula. La enfermedad perdura de por vida, generalmente indiagnosticada si es poco intensa, y diagnosticada, si media o grave. Aparato ocular. Los síntomas clínicos de xeroftalmía, con ser frecuentes, no son constantes en el SS1, y faltan en cerca de la mitad de los pacientes, unas veces porque las dacrioglándulas no se afectan y otras porque lo hacen ligeramente y no tienen manifestación clínica. La xeroftalmía se inicia clínicamente por la hiposecreción de las dacrioglándulas acuoserosas (glándulas lacrimales principal y accesorias), lo que se manifiesta en los casos leves o grado 1 por moderada inestabilidad de la película lacrimal, irritación ocular, enrojecimiento, escozor, sensación de arenilla y crisis de visión borrosa. A veces puede haber crisis de lagrimeo paradójico por irritación de la superficie ocular (Montero et al 1986). En los casos medios o grado 2 hay ruptura fácil de la película lacrimal, rivi lacrimales escasos con detritos abundantes, filamentos y placas muco-epiteliales precorneales, erosiones epiteliales corneales y conjuntivales (figura 4-1), y ligera hipostesia corneal. En los casos graves o grado 3 hay ulceraciones corneales estromales, vascularización y opacificación corneal, y muy raramente, perforación ocular y ceguera. La hinchazón de las glándulas lacrimales es rara. El cuadro clínico cursa espontáneamente con altibajos, de forma que las mejorías y peorías espontáneas son achadas a veces por los pacientes a tratamientos o circunstancias sin relación. La tendencia del cuadro es a progresar paulatinamente, pero esto se hace unas veces con gran lentitud y otras con gran rapidez.
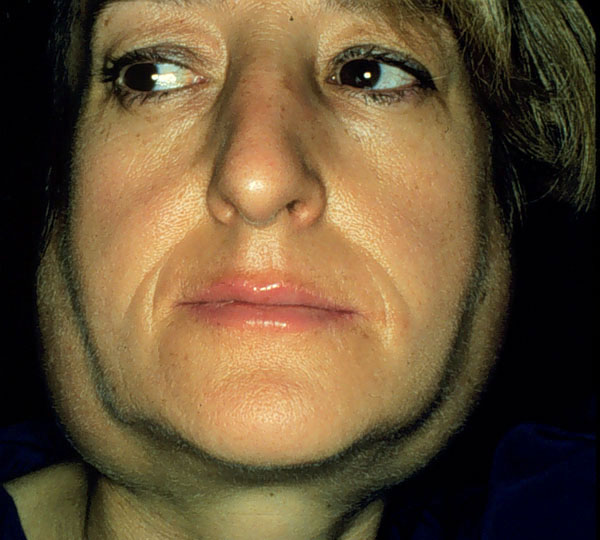
La afectación de las otras dacrioglándulas (mucosas, lípidas) se manifiesta más tardíamente, lo que ha hecho pensar a muchos investigadores que es secundaria a la disminución de la secreción serosa. Sin embargo, esto no es así, y en casos medios y graves, acaba presentándose aunque se mantenga un buen suministro acuoso al ojo con lágrimas artificiales. El deterioro de las glándulas mucíparas se manifiesta por los mismos síntomas ya citados. El deterioro de las glándulas del borde palpebral se manifiesta principalmente por blefaritis. Los signos de sequedad ocular han recibido diversos nombres según su fanerismo e intensidad : keratitis filamentosa (Leber 1882), keratitis sicca (Duke Elder 1930), keratoconjunctivitis sicca (Sjögren 1933), ojo seco (de Roetth 1950), etc. y algunos autores, confundiendo el signo con el síndrome, han aplicado aquellos nombres como sinónimos de síndrome de Sjögren. Aparato digestivo. La xerostomía por insuficiencia secretoria de las glándulas salivales mayores y menores se manifiesta en las formas ligeras por pastosidad de la saliva y por polidipsia provocada por la necesidad de humedecer la boca. Las formas medianas presentan fisuras o úlceras linguales, labiales y bucales, caries dental y trastornos del gusto (disgeusias). Las formas graves tienen imposibilidad de tragar y hablar o dolor al hacerlo, ulceraciones mucosas y estomatitis infecciosas; las infecciones más frecuentes son las candidasis, especialmente en pacientes que se tratan con glucocorticoides. Las glándulas salivales más afectadas son las serosas, por lo que entre las mayores, las más alteradas son las parótidas y las menos, las sublinguales. A veces aparecen episodios de hinchazón parotídea (adivas, paperas) (figura 4-2) de origen inmunológico. Estos episodios son distintos a los provocados por sialolitos, que pueden obstruir los conductos glandulares y provocar cuadros intercurrentes o permanentes de infarto glandular, con dolor local o adenitis antibiótico-resistentes, que a veces tienen que ser intervenidos quirúrgicamente.
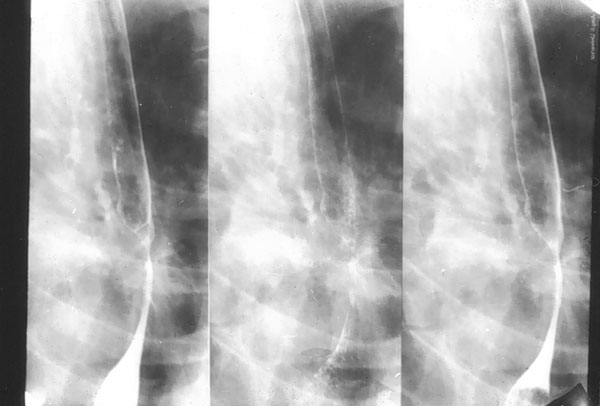
Otras manifestaciones digestivas de origen exocrino directo o indirecto son la dismotilidad esofágica (figura 4-3) e intestinal, achalasia, hipoclorhidria o aclorhidria, gastritis crónica atrófica, enzimodeficiencia pancreática, disfunción biliar y cirosis hepática.
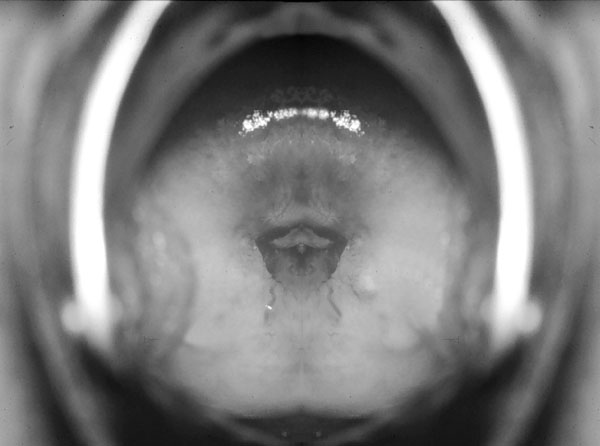
Aparato respiratorio. Su insuficiencia exocrina puede manifestarse por sequedad nasal, anosmia, epistaxis, faringolaringitis, disfonía, bronquitis, tos, respiración superficial y pneumonía. Aparato génito-urinario. Alrededor del 15 % de pacientes de SS diagnosticados fehacientemente tienen síntomas genitales y urinarios como sequedad o prurito vaginal, colpitis (figura 4-4) coito doloroso (dispareunia) y polaquiuria (Beiras 1947).
En el 20 % de los SS1 se encuentran infiltraciones linfocitarias intersticiales del riñon, que llevan a la atrofia tubular y a la fibrosis; muchos de ellos tienen acidosis tubular renal. Piel. La xerodermia por sequedad de las glándulas eccrinas y apocrinas de la piel se da en el 25% de los SS1 diagnosticados. Otras manifestaciones clínicas posibles son eczema, prurito, dispigmentosis, disminución del panículo adiposo, alopecia y fotoalergias. En el 70% de los SS1 se encuentran depósitos intraepidérmicos de IgG en piel clínicamente sana, mientras que iguales depósitos sólo aparecen en el 10% de los SS2. Manifestaciones no exocrinas. En el SS1 es relativamente frecuente encontrar lesiones orgánicas derivadas de la infiltración linfocitaria y de la vasculitis. La infiltración linfocitaria, que afecta típicamente a las glándulas exocrinas, alcanza también a numerosos órganos, sobre todo al sistema linfático. El sistema linfático manifiesta en un 20% de los SS1 infarto de los ganglios del cuello, axila, ingle, etc. El estudio histopatológico de estos ganglios los muestra llenos de linfocitos, plasmocitos y reticulocitos, que distorsionan y alteran la estructura interna del ganglio, e incluso aparecen por fuera de la cápsula ganglionar. A veces, la diferenciación entre lesión benigna y maligna es dificil La vasculitis produce lesiones perivasculares que alcanzan la piel, nervios, músculos, articulaciones, etc. Se calcula que el 15% de los SS1 tienen fenómenos de vasculitis. Como resultado de lo anterior, se describen en el SS1 frecuentes manifestaciones no exocrinas : nefritis intersticial (en el 20% de los casos), manifestaciones cutáneas en forma de petequias, dispigmentosis o necrosis (15%), manifestaciones neurológicas en forma de meningitis o trastornos psiquiátricos (15%), tiroiditis crónica de Hashimoto (5%), fenómeno de Reynaud (20%), pneumonitis intersticial, fibrosis pulmonar o pneumonía nodular cavitaria (10%), hepatomegalia o cirrosis biliar (10%), miositis (5%) y discrasias hemáticas como hipergammaglobulinemia, macroglobulinemia, crioglobulinemia o púrpura tromboocitopénica (Velilla et al 1975, García Bragado et al 1980). Se han publicado asociaciones infrecuentes en las que se desconoce si la coincidencia es casual o sindrómica : enfermedad de Behçet (Ramírez et al 1973), hipoovarismo (Ayala et al 1979), etc. Especial mención merece la degeneración de los infiltrados linfocíticos en pseudolinfomas (denominación dada por Talal et al 1964 a la situación intermedia y dudosa entre el simple infiltrado benigno y el linfoma) y linfomas diseminados no Hodgkin (Talal et al 1964) e inmunoblastosarcomas (Robin et al 1977, Aiwaza et al 1979). El 5% de los pacientes con SS1 definidamente diagnosticado desarrollan un linfoma maligno a partir de los linfocitos B; todos estos casos tienen anticuerpos anti-SS-A o anti-SS-B. Otros signos de riesgo de malignización son la existencia de linfoadenopatías, esplenomegalia y paperas sjögrénicas, También tienen más probabilidad de desarrollar linfomas los pacientes cuyas parótidas han sido tratadas con irradiación, pero como el linfoma suele aparecer fuera de la parótida, se supone que ello no se debe a la irradiación, sino a que ésta se aplicó a los enfermos con mayor reacción linfocítica. Es opinión de algunos autores que las drogas citotóxicas también aumentan el riesgo de linfomas. También es de destacar que los pacientes con SStienen ciertas particularidades alergológicas. Por ejemplo, si se intaura un tratamiento sistémico con sales de oro, con penicilamina o con levamisole, es importante saber que los pacientes con SS1 tienen mayor incidencia de reacciones alérgicas a estas drogas, mientras que los pacientes con SS2 no son más propensos a ellas que cualquier paciente con artritis reumatoide u otras colagenosis
Asociaciones Se ha encontrado gran número de manifestaciones patológicas asociadas al SS, de las que algunas son manifestaciones no exocrinas del SS1, otras son manifestaciones sindrómicas del SS2, y otras son asociaciones accidentales de interpretación dudosa. Así, se han publicado casos de SS asociados a alacrimia congénita (Lisch 1937 Coverdale 1948), anemia aplástica y linfoma (Fye et al 1980), anemia aplástica y timoma (Krieger et al 1981), anemia perniciosa (Houwer 1927), anhidrosis (Arruga 1934, Beiras 1947), aquilia (Fornés 1958), crioglobulinemia (Shah et al 1978, García Bragado et al 1980, Jouet et al 1980, Clerc et al 1980), dermatitis herpetiforme (Fraser et al 1979), diabetes mellitus (Sjögren 1935, Guzzinatti 1954, Palomar 1956), espondilitis anquilosante (Montero et al 1988a), eczema (Houwer 1927), enfermedad de Raynaud (Moutsopoulos et al 1979), esclerodermia (Alarcón 1974, David et al 1980), estenosis esofágica (Burkert et al 1980), fasciitis eosinofílica (Kaplinsky et al 1980), fibrositis intersticial pulmonar (Strimlan et al 1976, Brune et al 1976, Grange et al 1979, Chambard 1980, De Cremoux et al 1980), glomerulopatías y tubulopatías renales (Kahn 1962, Shearn etal 1961, Tu et al 1968, Flynn et al 1976, Moutsopoulos et al 1978a), hepatopatías primitivas (Alarcón et al 1973, Vogel et al 1980, Hamlyn et al 1980, Pennec et al 1981), hiperlipoproteinemia (Reinertsen et al 1980), hipotiroidismo e hipoovarismo (Ayala et al 1979), lupus erythematosus systemicus (Alarcón et al 1974, Alspaugh et al 1979), macroglobulinemia de Waldenström (Talal et al 1967, Velilla et al 1975), manchas cutáneas (Riaza 1960), periarteritis nodosa (Meur 1977), poliartritis reumatoide (Bergere 1980), polimiositis (Meur 1977), púrpura trombocitopénica (Meur 1977), rosácea (García Miranda 1948), sarcoidosis (Jones et al 1957), sarcoma de Kaposi (Patri et al 1980), sequedad vaginal (Gougerot 1925, Beiras 1947, Fornés 1958), uveítis (Díaz Valle et al 1996), síndrome de Behçet (Ramírez et al 1973, Hamza et al 1980), síndrome de Reynolds (Trotta et al 1980), tiroidopatías de Hashimoto (Wechsler et al 1976, Meur 1977), Moutsopoulos 1980), etc. Algunas asociaciones se hacen a oftalmoxerosis no claramente definidas como SS, como la oftalmoxerosis con placas mucoides, asociada a colitis ulcerosa, eritema nodoso, psoriasis o reticulosis benigna (Fraunfelder et al 1977).
CRITERIOS DIAGNÓSTICOS Todo presunto paciente de SS debe ser sometido a un cuidadoso interrogatorio, y si por él y por las exploraciones de orientación se sospecha el diagnóstico de SS, deberá ser atendido al menos por un oftalmólogo y por un reumatólogo. El primero estudiará el problema ocular y si procede, impedirá el desarrollo de las lesiones de superficie ocular. El segundo asistirá al paciente en su campo específico y detectará, si la hubiere, una neoplasia linfoide. Ambos recabarán la analítica y exploraciones necesarias, y pedirán si fuese menester el concurso del estomatólogo, ginecólogo, pneumólogo, inmunólogo, etc. Hay diversos criterios diagnósticos del SS. Se dice que un SS es "definido" cuando reúne todos los requisitos mínimos según un determinado criterio, y que es "probable" cuando le falta alguno de esos requisitos. Los criterios mínimos para el diagnóstico de un SS1 exigen la existencia de una KCS y la comprobación de una agresión del sistema inmunitario a las glándulas exocrinas. Para la definición de un SS2 es necesario que al anterior diagnóstico se añada una conectivopatía crónica autoinmune. La KCS se puede diagnosticar con los criterios de Pisa, expuestos en el capítulo 23, que exigen la positividad de la prueba de Schirmer y de la tinción vital de la superficie ocular, y potestativamente del descenso de una proteína lacrimal (lisozima, lactoferrina o IgA) y el ascenso de la osmolaridad. La participación de las glándulas exocrinas se determina mediante biopsia de glándula labial, por su fácil accesibilidad. El diagnóstico de la conectivopatía se hace por la clínica y por la analítica serológica Pruebas más sofisticadas pueden corroborar o no el diagnóstico, pero no lo excluyen aunque sean negativas. Por otra parte, la negatividad o positividad de las pruebas inmunológicas típicas (antígenos HLA y anticuerpos característicos) tienen sólo valor orientativo, pues al no ser absolutamente típicas, ni excluyen ni indican con certeza el diagnóstico de SS. Estas pruebas hematológicas se exponen en el capítulo 32. Los criterios diagnósticos más usados hasta el presente son: Criterio de Copenhague (Manthorpe et al 1986). Exige para definir un SS1 la existencia de 1) xeroftalmía diagnosticada al menos por 2 de las siguientes 3 pruebas: prueba de Schirmer inferior a 10 mm, tisc/but inferior a 10 sec, y tinción con rosa de bengala superior a 4 según el tanteo de van Bijsterveld. Y 2) xerostomía diagnosticada al menos por 2 de las siguientes 3 pruebas: sialometría de secreción no estimulada inferior a 1'5 ml/15min, sialoscintigrafía que muestras secreción deficitaria, y biopsia glándulolabial que muestre infiltración linfocitaria positiva. Criterio Griego (Moutsopoulos et al 1979, Skopouli et al 1986). S1 definido: 1) Dos de las 3 siguientes manifestaciones: Xeroftalmía con prueba de rosa bengala positiva, o xerostomía o paperas recurrentes. 2) Biopsia glándulolabial positiva. 3) Ausencia de otras enfermedades autoinmunes sistémicas. SS1 posible: 1) Una de las 3 primeras manifestaciones anteriores, y 2) biopsia glandulolabial positiva. SS2 definido: 1) xerostomía con o sin xeroftalmía. 2) artritis reumatoide y 3) biopsia glandulolabial positiva SS2 posible: 1) artritis reumatoide u otra conectivopatía autoinmune, 2) xeroftalmía subjetiva o con prueba de rosa bengala positiva y 3) biopsia glandulolabial positiva sin xerostomía Criterio de San Diego (Fox et al 1986). Excluye del diagnóstico de SS a los pacientes con linfoma preexistente, sarcoidosis, síndrome de inmunodeficiencia adquirida (sida) o enfermedad del transplante. En los demás, se diagnostica como SS definido o como probable a los que tienen 4 ó 3, respectivamente, de las sigiente determinaciones: 1) xeroftalmía diagnosticada con la prueba de rosa bengala y/o fluoresceína, 2) xerostomía, 3) biopsia glandulolabial positiva, y 4) anticuerpos anti-SS-A y antiSS-B, o factor reumatoide positivos
PRUEBAS DIAGNÓSTICAS Las pruebas diagnósticas son expuestas en la parte 4 de este libro. Sólo citaremos aquí algunas particularidades del SS.
Cuantificación lacrimal La secreción lacrimal basal está disminuida desde el inicio de la enfermedad. Al principio esto puede ser difícil de establecer con una pruebas de ambiente clínico como es la prueba de Schirmer o la prueba de secreción basal de Jones. La secreción refleja también disminuye, según es observación general con la prueba de Schirmer y según comprobó De Haas 1964a tras inhalación de vapor de amoniaco. La prueba basal de Jones es muy útil La secreción farmacoinducida tambien tiene una respuesta menor a la normal en el SS, pero esto también ocurre en la queratitis filiforme y en otras oftalmoxerosis, tanto a la estimulación con pilocarpina (Betsch 1928, Hauer 1931, De Haas 1961b, 1964a) como con neostigmina y metilcolina (De Haas 1964a)
Análisis de proteínas de la lágrima Cuando comienza a decaer la función de la glándula lacrimal, aún antes de que aparezcan signos y síntomas evidentes, los valores de lisozima y lactoferrina descienden y los de ceruloplasmina ascienden. Los valores de IgA se disminuyen con menos intensidad.
Osmolaridad de la lágrima La primera referencia a que el ojo seco se asocia a una lágrima muy salina lo recogemos en Galeno (siglo II AD) cuando en el tomo XIV de sus obras, en E Iatroz., dice que el cuadro clínico de la xeroftalmía es úlcera angular, aspereza, prurito, párpados rojos y flujo de lágrima salada y nitrosa. La osmolaridad de la lágrima normal es de 300-310 mOsm/litro y en los síndromes de ojo seco asciende a 320-340 mOsm/l. Mastman et al 1961 encontraron que la osmolaridad de la lágrima normal es equivalente a una solución cloruradosódica del 9'53 por mil y que en 22 pacientes de KCS era del 9'70 por mil. Sullá et al 1992 encontraron valores medios de 307 mOsm/l en personas normales y 339 mOsm/l en pacientes con ojo seco. La osmolaridad de un líquido puede medirse de varias formas, pero la generalmente usada en medicina es por el punto de congelación, que desciende tanto más cuanto mayor el la salinida del líquido, en este caso, de la lágrima. La osmolarimetría de la lágrima es prueba de uso corriente en clínica dacriológica especializada. Se necesita una muestra mínima de 0'1 m l La osmolaridad de la lágrima en el SS y demás ojos secos suele estar aumentada. La razón de esto puede ser cuádruple:1) Aumento primario del componente salino del secretado, sea mayor o menor la producción acuoserosa. 2) Disminución del volumen del mar lacrimal con mantenimiento de su superficie de contacto con el aire, con el consiguiente mayor valor relativo de la evaporación. (3) Menor producción acuoserosa, aunque sea normoosmolar, lo que conlleva una menor tasa de renovación del mar lacrimal y por tanto permite que la evaporación actúe más tiempo sobre la misma lágrima. 4) Disminución de los protectores físicoquímicos de evaporación, tales como la capa lípida de la película lacrimal y la dispersión de la mucina. Cuando la evaporación es normal y la osmolaridad alta, se supone que la capa lípida es normal, y que hay una baja tasa de renovación por escasa producción acuoserosa. Cuando la osmolaridad sube de forma aguda a niveles similares a los de ojos secos (330 y 360 mOsm/l), Vergés et al 1985 han demostrado en un modelo animal que se produce el vaciado de la mucina de las células caliciformes y se inhiben sus mecanismos de síntesis; este vaciado de mucina es máximo entre las 6 y 8 horas de la exposición a las soluciones hiperosmolares, por lo que la tinción con PAS no identifica las células caliciformes y hace pensar que han desaparecido. Sin embargo, cuando la exposición a la hiperosmolaridad se prolonga, el descenso de células caliciformes es real (Vergés et al 1986, 1990). Para más información sobre la osmolaridad ver los capítulos 30 y 39.
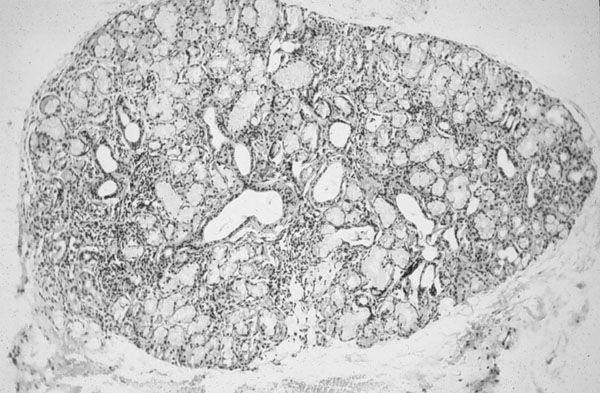
Histopatología La biopsia de las glándulas lacrimales acuoserosas y salivales (figura 4-5) muestra que las lesiones del SS afectan tanto a la glándula principal como a las accesorias (Sjögren 1940). El parénquima secretorio aparece atrofiado. En el estroma circundante hay edema mucoso, degeneración fibrinoide, hialinización y esclerosis del tejido conjuntivo. La infiltración linfocitaria es constante (Escande 1977). Los linfocitos B forman acervos perivasculares, mientras que los linfocitos T, más escasos están dispersos (Belfort et al 1980). También se encuentran células vacuoladas en los espacios interacinares (Sarada 1980)
Arenas et al 1996que el diagnóstico de SS2 se hace sobre los síntomas (síntomas de ojo y boca seca, artritis reumatoide, etc), prueba de Schirmer I e infiltración linfoplasmocitaria de la biopsia conjuntival. Para más información de biopsias glandulares ver los capítulos 33 y 34.
TRATAMIENTO Para el tratamiento del síndrome de Sjögren, se expone en la sección de tratamiento. Sólo citamos aquí la posibilidad en un futuro de hacer un tratamiento preventivo. Se basa ello en los estudios de Haneji et al 1997, quienes han comprobado en ratones con SS1, que la inyección intravenosa de un recombinante de la a-fodrina proteje a los animales de la aparición de las típicas lesiones glandulares del SS. Aún es pronto para saber si esto será aplicable a humanos. |