CAPÍTULO 15 OJOS SECOS POR DEFECTOS CONGÉNICOS Fabián Ponce |
| AGENESIA DE GLÁNDULAS LACRIMALES Las glándulas lacrimales se forman por una invaginación en el mesodermo subyacente de las células epibláticas que van a formar la conjuntiva. Esto se inicia en el embrión de 24 mm (43 días de desarrollo). Los acinos secretorios se forman durante el 4.º mes de desarrollo. La glándula es capaz de secretar basalmente, sin inervación, desde unas semanas antes de nacer, de forma que un feto prematuro no tiene sequedad ocular extrema. La inervación parasimpátosecretora no se completa hasta el nacimiento o unas semanas después, por lo que la secreción refleja no aparecerá hasta después del nacimiento. Cuando las glándulas o su sistema de inervación no se desarrollan, la persona no tendrá lacrimación o la tendrá reducida a la cuantía en que aquellas se desarrollaron. La falta total de secreción acuoserosa es incompatible con la conservación de la transparencia corneal. Con el nombre de alácrima o alacrimia se ha designado la falta de lágrima, sea por falta parcial de glándulas acuoserosas, en cuyo caso falta la secreción basal y la córnea se opacífica, o sea por falta de inervación, en cuyo caso hay un tipo de secreción basal que permite una película lacrimal inestable. La mayoría de los autores (p.ej. Sjögren et al 1950) denominan alacrimia a la ausencia de lagrimeo reflejo y emocional con conservación del basal. La alacrimina puede asociarse a asialia y a otras malformaciones como agenesia de los puntos lacrimales (Caccamise et al1981).
SÍNDROME DE BONNEVIE-ULLRICH El síndrome de Bonnevie-Ullrich es un síndrome congénito con edema linfangiectásico de pies, manos y cuello presente ya al nacer, con piel hiperelástica, hipotrofia muscular y, a menudo, parálisis de pares craniales, cataratas congénitas, epicanthus, etc. A veces faltan las glándulas lacrimales y la carúncula. Afecta más a mujeres que a varones en proporción 4/1. No se ha establecido un factor hereditario
ANIRIDIA La anomalía congénita irido-conjuntival de Bietti fue publicada por este autor en 1963 quien vio 5 casos de una dysgenesia mesodermalis asociada a xerosis conjuntival. Los pacientes tenían diversas malformaciones iridopupilares asociada a ausencia de células caliciformes de la conjuntiva, con xerrosis ocular y manchas de Bitot. Posteriormente se han aportado nuevos casos (Meyer 1965, etc.) La aniridia frecuentemente coexiste con alteración de las células madre limbales e invasion del epitelio corneal por células de fenotipo de conjuntiva, que "conjuntivalizan" el epitelio corneal. (Tseng et al 1996).
DISPLASIA ECTODÉRMICA ANHIDRÓTICA La displasia ectodérmica anhidrótica, también conocida como síndrome de Siemens 1925, es una enfermedad congénita familiar autosómica recesiva, que afecta en el 95% de los casos a varones. Se caracteriza por una ausencia más o menos completa de glándulas sudoríparas y sebáceas, e hiperqueratosis folicular con hipotricosis y malformaciones dentales. A veces se asocia con síndrome de Horner, asialia, etc. En el aparato lacrimal se ha descrito, aunque infrecuentemente, la alacrimia acuoserosa, (Arruga 1934 Thurnam 1948) y la agenesia de glándulas de Meibomio (Holly et al 1977b)
DISPLASIA ECTODÉRMICA HIDRÓTICA La displasia ectodérmica hidrótica ocasionalmentre puede dar síntomas y signos oculares. Adenis et al 1979 ha descrito la participación de una secreción acuoserosa cuantitativamente normal, y abundancia de mucus filante, lo que produce un tisc/but bajo.
SÍNDROME DE CURTIUS El síndrome de Curtius fue descrito por este autor en 1925. Es una displasia ectodermica hidrótica de la piel con alteraciones endocrinas (gigantismo), y malformaciones cefálicas (hipertelorismo) y oculares (catarata, hipotricosis de pestañas y cejas, nystagmus, degeneración tapetorretiniana), a las que a veces se añade hiposecreción lacrimal.
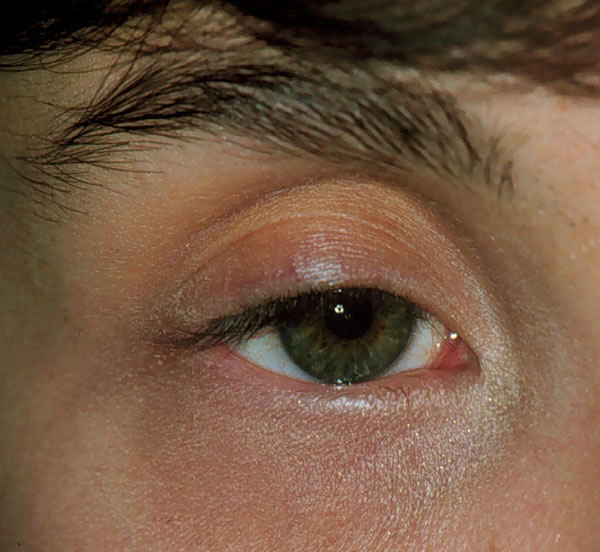
AGENESIA DE GLÁNDULAS DE MEIBOMIO Es un defecto raro. Casi siempre se da asociado a otras malformaciones, como hipoplasia tarsal o hipoplasia palpebral. Cuando ocurre, con frecuencia se acompaña de agenesia de folículos pilosebáceos, y por tanto de pestañas y sus glándulas asociadas. Nosotros lo hemos visto asociado a algunos síndromes de epicanto-blefarofimosis y a síndromes de hipoplasia del primar arco branquial (figura 14-1).
SÍNDROME DE RILEY-DAY Síndrome de Riley-Day. Ver capítulo 12. |