|
DEFINICIONES
Retinoblastoma: tumor maligno de las células primitivas de la retina
sensorial, que aparece en los primeros años de vida del niño.
Leucocoria: reflejo pupilar blanquecino.
GENERALIDADES
(1-8)
• Primer tumor intraocular en la infancia.
• Incidencia: Uno de cada 14.000-23.000 nacidos vivos. Para J.A. Shields 1
de 15.000.
• Edad media al diagnóstico: bilaterales 12 meses; unilaterales 23 meses.
• No hay preferencia de sexo ni de raza.
• 25-30 % son bilaterales.
• Puede haber regresión espontánea.
• Actualmente sobreviven un 95%, y en las mejores estadísticas se consigue
hasta un 75% de salvación del ojo en los grados V de RB.
¿Es ético intentar salvar un globo ocular, por todos los medios posibles?
Nos lo planteamos:
1 Muchas veces la familia quiere que se le conserve y nos obliga a terapias
conservadoras.
2 Puede aparecer un tumor en el contralateral a los 4-5 años de enuclear un
ojo.
Es muy importante la experiencia de un equipo multidisciplinario en este tipo
de patologías.
Correlación anatomoclínica
La leucocoria es muy manifiesta cuando el tumor adquiere un tamaño
considerable. Se presenta en un 60-70% de las estadísticas consultadas (1-3,5).
El foco del retinoblastoma en sus inicios, pasa desapercibido. Cuando es
intrarretiniano, se presenta como una tumoración blanca, nacarada, que emerge
de la retina sensorial.
Inicialmente es pequeño y transparente, siendo de muy difícil
visualización, tanto clínica como macroscópica.
Suele al crecer volverse opaco y al elevarse se convierte en la masa blanca
típica que conocemos (figuras 1a y 1b).

Figura 1a. Diversos tipos de retinoblastoma.
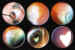
Figura 1b. Retinoblastomas en la Ora Serrata adoptando múltiples
formas.
Cuando se desarrolla se vasculariza estableciéndose una circulación
(aferente y de drenaje) de los vasos retinianos (2,9).
Puede dar siembra vítrea llegando en su diseminación a simular una
endoftalmitis (2,5)
En casos extremos afecta a la cámara anterior dando hipopión, sinequias,
hiphema y glaucoma neovascular secundario (10-12). Puede haber siembra tumoral
en la malla trabecular (13-16).
Si crece hacia el vítreo, la retina permanece estable a su alrededor y la
forma tumoral se denomina endofítica.
Hay vasos dilatados y tortuosos en la base del tumor. El vértice es
avascular aparentemente, aunque en la A.F.G. se demuestra microcirculación en
todo el tumor y pérdida de contraste (2,13).
Si produce un desprendimiento de retina con áreas sólidas y bolsas de
fluido subretiniano alrededor se denomina exofítico.
Hay células tumorales en el líquido subreti niano.
En la neoformación se suelen apreciar áreas de necrosis (amarillentas) y
calcificaciones. Los vasos retinianos están por encima del tumor.
La tumoración puede atravesar la membrana de Bruch y pasa a coroides
(11,12).
Muchas veces el patrón es mixto y dejándole evolucionar da un gran
porcentaje de glaucoma neovascular (2,5,9).
En rarísimas ocasiones es infiltrante difuso, asemejándose a una
afectación inflamatoria ocular. Puede verse la celularidad pseudoinflamatoria
en retina, vítreo o en cámara anterior (pseudohipopión) (12-15). Se da en
niños mayores (5-7años). La ecografía, el TC y la RM fallan habitualmente en
el diagnóstico y sólo la citología y la biopsia los diagnostican. Conviene
enuclear antes de las 48 horas por el peligro de extensión o metástasis
(14-25).
CLÍNICA
La leucocoria es la primera manifestación del retinoblastoma.
El estrabismo es el 2.º síntoma, endotropía en un 15%, exotropía 5%.
Hyphema espontáneo, heterocromía de iris rubeo sis, hipopión, uveítis,
glaucoma, celulitis orbitaria, exoftalmos, ojo rojo y nistagmo son otras
referencias de diagnóstico de la patología (1,2,4,8,13,19,26).
Los pacientes hemofílicos, leucémicos y con xantogranuloma juvenil pueden
dar síndromes de enmascaramiento (5,13,19,27) (figuras 2 a y b).

Figura 2a. Los síndromes de enmascaramiento son múltiples y
variados.
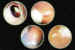
Figura 2b. Los retinoblastomas con siembra vítrea periféricos
pueden producir síndromes de enmascaramiento.
En niños mayores, los retinoblastomas infiltrantes difusos y unilaterales no
hereditarios aparentan una enfermedad inflamatoria produciendo confusión en el
diagnóstico y tratamiento (13-28). Hay peligro de extensión y diseminación si
se realizan múltiples pruebas diagnósticas y biopsias (11,17,18,29,30,31).
Los casos avanzados son de mal pronóstico; se acompañan de glaucoma,
extensión por el nervio óptico y órbita, afectación del cerebro por
contigüidad o vía espacio subaracnoideo y metástasis (10-12,15,16,29,32). No
tiene tanta importancia la afectación de la lámina cribosa o la afectación
coroidea si no es masiva (12,13,33).
Conceptos erróneos
• No se da retinoblastoma en ojos microftálmicos o con persistencia de la
vasculatura fetal (falso) (28, 34-36).
• No hay RB en el recién nacido (falso) (37).
• No se da el RB con cataratas congénitas (falso) (38,39).
• No aparecen retinoblastomas en la edad adulta (falso) (16,40-42).
• La radiación produce un 30% de segundos tumores en los afectados
genéticamente a los 30 años (falso) (17).
LA ÓRBITA, EL RETINOBLASTOMA Y EL MÁS ALLÁ
La extensión del retinoblastoma fuera de las cubiertas oculares, a la
órbita por contigüidad, al nervio óptico o al sistema nervioso central y las
metástasis hemáticas o linfáticas muestran en las estadísticas un
pronóstico vital muy malo (11,29).
Sin embargo ya se han publicado artículos con resultados sorprendentes
combinando cirugías agresivas, quimioterapias y radioterapias en casos de
afectación orbitaria (43,44).
Afortunadamente la existencia de pacientes con histología adversa ha
disminuido en estos últimos años pero precisan nuevas quimioterapias y
radioterapias sobreañadidas en zonas afectas que habitualmente no se utilizan.
Están en fase experimental por el momento la radioterapia con protones, gamma
knife y aceleradores lineales tridimensionales (45-49).
Quimioterapias intratecales e incluso autotransplante de médula ósea en
casos ya muy desesperados utilizan múltiples y nuevas drogas y agentes
inmunosupresores (51-53).
Si añadimos el factor genético a todos estos tratamientos estamos
favoreciendo la aparición de segundas neoplasias y tumores iatrogénicos.
Siempre debemos pensar en los tumores de línea media cerebral, retinoblastomas
trilaterales (r.b. bilateral+pinealoblastomas) etc. (7,18,21,29,53-56).
La exenteración orbitaria ha caído un poco en desuso, pero realizada con
indicaciones precisas permite obtener muy buenos resultados, en aquellos casos
en que los riesgos genéticos llevarían al paciente a un cúmulo de desgracias
sumativas (33,43,52).
Por suerte son pocos los niños en nuestras casuísticas que desarrollan
estos procesos y los avances en técnicas focales permiten mejorar la ca lidad
de vida y evitar complicaciones (6,37,57-59).
MANIFESTACIONES MUY INFRECUENTES
Retinoblastoma con regresión espontánea
1% de las casuísticas (1,2,4,5,8,13,19).
Tumor inactivo. Se produce necrosis isquémica, calcificación,
proliferación de células del epitelio pigmentario retiniano, células gliales,
e incluso osificación.
Muchas ptisis bulbi con grandes inflamaciones previas en niños menores de 5
años pueden deberse a una regresión espontánea del tumor que pasa
desapercibido. Debe sospecharse siempre cuando ocurra.
Retinocitoma
Variante benigna del retinoblastoma. Es el 2% de las casuísticas de esta
patología (59,63).
Según Zimmerman (63) tienen células sin características de malignidad,
aunque aparentan rosetas y fleurettes, bien diferenciadas, en un tumor
calcificado con áreas traslúcidas, muy parecido al grado 3 de regresión del
retinoblastoma. Hay proliferación del epitelio pigmentario. Los vasos
retinianos penetran en la masa tumoral, con algunos neovasos en la AFG. Hay
descritos casos de crecimiento y malignización (60,64), por lo que hay que
vigilarlos con mayor frecuencia de lo habitual. Un retinocitoma tiene la misma
trasmisión genética que los retinoblastomas con todo lo que ello implica.
Retinoma
Para J.A. Shields (2), este término es demasiado general y significaría
cualquier tumor en la retina. En la literatura se asocia con los retinocitomas,
considerándose la misma entidad (59,60,62,64-66).
DIAGNÓSTICO DIFERENCIAL
(106)
Shields y col. (66) publican una serie de más de 100 pacientes con el
diagnóstico de sospecha de retinoblastoma. Tras estudiar los casos, se confirma
este diagnóstico en el 58% de los pacientes. En el resto, el diagnóstico final
fue de persistencia de vítreo primario hiperplásico (PVPH) en el 28%,
enfermedad de Coats en otro 16% y toxocariasis ocular en otro 16%.
Howard y Ellsworth (53) estudiaron 500 pacientes diagnosticados de
retinoblastoma. En el 53% de la serie hubo que cambiar el diagnóstico.
Encontraron un elevado porcentaje de casos de persistencias de la vasculatura
fetal, que fue catalogada en un principio como retinoblastoma. Otros
diagnósticos fueron fibroplasia retrolental, catarata posterior y uveítis.
Las entidades que pueden confundirse con cierta frecuencia son (figuras 3 y
4):
• Persistencia de la vasculatura fetal (P.V.F).
• Retinopatía de la prematuridad grado V.
• Enfermedad de Coats.
• Toxocara ocular.
En alguna ocasión es difícil el diferenciarlos:
• Xantogranuloma juvenil masivo.
• Facomas retinianos.
• Pars planitis con gran actividad y banco de nieve con desprendimiento.
• Endoftalmitis metastásica.
• Meduloepiteliomas muy avanzados.
Raramente pueden ser confundidas:
• Coloboma del nervio óptico.
• Coloboma de polo posterior.
• Uveítis.
• Catarata.
• Fibras nerviosas mielinizadas.
• Displasia retiniana (enfermedad de Norrie).
PERSISTENCIA DE LA VASCULATURA FETAL (Persistencia del vítreo primario
hiperplásico)
Morton F. Goldberg en el LIV Memorial Edward Jackson (1997) propuso
definitivamente el cambio de nombre de la persistencia del vítreo primario
hiperplásico, por persistencia de la vasculatura fetal al considerar que se
integran en su definición y clasificación muchas más entidades de esta
compleja patología. Pasaran muchos años hasta que nos acostumbremos a esa
nueva denominación (PVF), pero tendremos que hacerlo porque la antigua (PVPH)
es incompleta.
Dentro del diagnóstico diferencial del retinoblastoma y otras causas de
leucocoria, uno de los cuadros que se deben tener en cuenta es la Persistencia
de la vasculatura fetal(PVF), antes llamada persistencia de vítreo primario
hiperplásico (PVPH). Descrito por primera vez por Reese (67), consiste en una
malformación congénita del vítreo primario que se presenta generalmente como
una placa de tejido conectivo fibrovascular retrolental. Esta estructura, que
puede contener tejido adiposo, cartílago y músculo liso (68), además de vasos
y fibroblastos, puede adherirse a la cápsula posterior del cristalino
conduciendo a la opacidad progresiva del mismo. Por otro lado, esta placa
fibrosa, puede extenderse lateralmente hasta unirse con los procesos ciliares
que sufrirán una elongación centrípeta. Un cristalino «intumescente» junto
con unos procesos ciliares proyectados anteriormente, producen el abombamiento
del iris que conduce a la presencia de una cámara poco profunda y al desarrollo
de glaucoma que característicamente se presenta asociado al cuadro de PVF.

Figura 3. 1. Toxoplasmosis, 2. Tuberculoma, 3. Facoma joven, 4.
Facoma calcificado, 5. PVF posterior, 6. Pliegue falciforme, 7 y 8 Fibras de
mielina, 9. Luxación de cristalino posterior, 10. Malformación con fibrosis
posterior, 11. Hamartoma combinado de retina y epitelio pigmentario, 12. Angioma
capilar papilar enmascarando un méduloepitelioma del nervio óptico, 13.
Morning Glory, 14. Coloboma de retina y coroides, 15. Estafiloma de papila, 16.
Coloboma de polo posterior.

Figura 4. 1. Leucocoria estrabismo y microftalmía en PVF, 2. y 3.
PVF reabsorbiendo el cristalino, 4. PVF con hemorragia apical, 5. Anomalía de
Peeters, 6. Displasia retiniana, 7. PVF tracto fibroso retrocristaliniano, 8.
Retinopatía del prematuro grado 5, 9. Endoftalmitis, 10. Retinopatía del
prematuro. Cámara anterior estrecha, 11. Cuerpo extraño intraocular vegetal,
produciendo endoftalmitis, 12. Catarata y atrofia de iris por radiación, 13.
Catarata y luxación de cristalino en síndrome de Marfan, 14 y 15. Pars
Planitis, banco de nieve y DR periférico, 16. Pars planitis, catarata
secundaria.
El 90% de los casos son unilaterales aunque con mucha frecuencia se pueden
encontrar en el ojo adelfo alteraciones menores en la regresión normal del
vítreo (69) como son la mancha de Mittendorf, opacidad nasal inferior en la
cápsula posterior del cristalino, recuerdo de la unión durante el período
fetal de la arteria hialoidea y la túnica vasculosa lentis, o una papila de
Bergmeister, remanente de la porción posterior de la arteria hialoidea.
Los casos bilaterales suelen verse en pacientes con otras anormalidades
sistémicas (68) (p.e. trisomía 13, labio leporino, paladar hendido,
polidactilia y microcefalia) y asocian generalmente afectación más severa del
polo posterior (grados variables de displasia retiniana).
Es posible encontrar PVF en el 3% de los recién nacidos a término, y en
algún grado, en el 95% de los prematuros (70). En niños menores de 36 semanas
de gestación, en más del 90% de los casos podemos hallar signos que implican
una regresión incompleta del sistema vascular hialoideo (71,72).
RETINOPATÍA DEL PREMATURO
(73)
Se da principalmente en prematuros menores de 28 semanas de gestación y de
1.500 gramos.
Etiología multifactorial. Oxígeno, hipoxemia, hipercapnia, factores
vasoproliferativos endoteliales, etc. (74,75).
El desarrollo del estadio III plus que es una neovascularización retiniana
periférica da tracciones vitreorretinianas, hemorragias y fibrosis. La
conclusión del cuadro es un desprendimiento de retina total.
Hay que realizar controles oftalmoscópicos neonatológicos entre la 4 y la 6
semana de edad cronológica o las 31-33 semanas desde la concepción, para
visualizar el desarrollo de la vascularización. Si ésta no se produce,
establecer un seguimiento periódico y los tratamientos pertinentes. Legalmente
hay que seguir al prematuro en las anteriores condiciones hasta los 6 meses y es
aconsejable posteriormente vigilar los estados cicatriciales toda la vida.
Siguen siendo frecuentes en nuestro país los casos que desarrollan el grado 5 y
cuando se refieren a centros especializados ya es tarde. Hay que hacer el
diagnóstico diferencial con la incontinentia pigmenti en niñas, con la
vitreorretinopatía exudativa familiar y el retinoblastoma principalmente en los
unilaterales (76).
TOXOCARIASIS
Los granulomas solitarios pueden parecer retinoblastomas aunque a menudo
muestran un centro traslúcido y hay signos de retracción sobre la retina. Se
localizan en la retina posterior o periférica (figuras 5 y 6). El nemátodo
provoca una inflamación localizada con bandas de tracción sobre la mácula.
Pueden producirse desprendimientos de retina y cataratas (77). Son raramente
bilaterales. Se diagnostican entre los 6 meses y los 6 años. Dan endoftalmitis
crónicas y el vítreo se vuelve opaco con lo que complican el diagnóstico. El
paciente suele tener historia de contacto con perros pequeños o hábitos de
comida en áreas sucias. La paracentesis de cámara anterior puede revelar
eosinófilos (78,79). El test de ELISA para toxocara puede ser positivo. La
tomografía computarizada, la ecografía y la resonancia diferencian las
entidades en casos límite.

Figura 5. Granulomas por toxocara.

Figura 6. Granulomas de polo posterior (toxocariasis).
ENFERMEDAD DE COATS
Microaneurismas en los vasos de la retina. Se dan con más frecuencia en los
cuadrantes temporales. Fácil de diferenciar con el retinoblastoma en estadios
iniciales, por sus microdilataciones y exudados amarillos intra y subretinianos
(5,80,81) (figuras 7 y 8).

Figura 7. Angiomatosis centrales y periféricas y enfermedad masiva
de Coats.

Figura 8. Enfermedad de Coats.
Cuando existe desprendimiento de retina exudativo con hemorragias y
desestructuración intraocular se precisan técnicas neuroradiológicas y AFG
para diferenciarlos. Se dan en pacientes hasta los 20 años pero los casos más
agresivos son en la primera década. Raramente es bilateral y no es hereditario
(4). La crioterapia, los diferentes tipos de láseres (figura 9), la diatermia
intraescleral y las vitrectomías con endoláser son procedimientos que se usan
con resultados poco satisfactorios en los grados avanzados.

Figura 9. Tratamiento con láser de angiomatosis periférica.
PARS PLANITIS
Sólo en casos extremos con inflamación generalizada o larga evolución por
desidia familiar se describen en la literatura dificultades para diferenciarla
del retinoblastoma. Hay síndromes de enmascaramiento diagnosticados como pars
planitis y ptisis confundidas con uveítis que eran retinoblastomas (82,83).
ENDOFTALMITIS METASTÁSICA
Secundarias a estreptococo, meningococo o estafilococo (83,84):
• Meningitis.
• Endocarditis.
• Sepsis abdominal.
TUMORES
Hamartoma astrocítico (facoma)
• Masa amarillo blanquecina que se calcifica. Si está aislada y muy
periférica puede confundirnos. Los tuberculomas se les parecen y alguna vez se
blanquean igualmente por inflamación y fibrosis (57,83,85,86). Los facomas se
asocian con la Esclerosis Tuberosa y raramente con las Neurofibromatosis. La AFG
y un estudio pediátrico o neurológico los diferencian claramente (86). Las
técnicas de imagen son muy útiles en las facomatosis.
Hemangiomas capilares
• Angiomatosis con gran componente exudativo intrarretiniano y subretiniano
(figura 10).
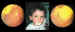
Figura 10. Leucocoria bilateral por angiomatosis de Von
Hyppel-Lindau.
• Herencia autosómica dominante. Hay que vigilar las manifestaciones
intracraneales en los Von Hippel Lindau y la patología renal (83,84).
• En los hemangiomas capilares es difícil confundirse pero también hay en
la literatura referencias. En nuestra casuística (85) tenemos un caso publicado
de asociación con meduloepitelioma de nervio óptico. El angioma era
blanquecino y el diagnóstico del meduloepitelioma se hizo por anatomía
patológica (vía neuroquirúrgica).
Hamartoma combinado de retina y epitelio pigmentario retiniano
• Masa ligeramente elevada de color gris-verdosa, que afecta al polo
posterior, incluye mácula y papila y se funde con retina normal (86,87).
• Tortuosidad vascular.
• Tumor benigno poco frecuente.
• Lesión congénita raramente evolutiva ,de crecimiento lento.
• Unilateral sin predominio de sexo.
• Aspecto funduscópico diagnóstico.
• Problemas de diagnóstico diferencial con tumores malignos y asociación
con NF2.
• Proliferación desorganizada de elementos gliales, vasculares y
epiteliales pigmentados.
• CAPA EXTERNA: EPR hipertrofiado.
• CAPA INTERNA: retina engrosada con disposición normal de sus capas.
• Tortuosidad vascular y tracción por membrana epirretiniana asociada.
BIBLIOGRAFÍA
- Devesa SS. The incidence of retinoblastoma. Am J Ophthalmol 1975; 80:
263-265.
- Shields JA. Retinoblastoma. In Guyer DR, Yannuzzi LA: retina, vitreous and
macula. Philadelphia, WB Saunders Co; 1999; 99: 1.139-1.150.
- Bechrakis NE, Bornfeld N, Schuler A, Coupland MBS, Henze G, Foerster MH.
Clinipathologic features of retinoblastoma after primary chemoreduction. Arch
Ophthalmol 1998; 116: 887-893.
- Spalton DJ, Shilling JS. The retina. Atlas of Clinical Ophthalmlogy. In
Spalton DJ, Hitchigs RA, Hunter PA. Wolfe Publishing. Londres 1994; 14:
14.2-15.3.
- Gallie B, Moore A. Retinoblastoma. In Taylor D: Paediatric Ophthalmology.
Blackwell Sciance Corp, 1996; 42: 519-536.
- Mahoney MC, Burnett WS, Majerovics A, Tanenbaum H. The epidemiology of
ofthalmic malignancies in New York State. Ophthalmology 1990; 97: 1.143-1.147.
- Sanders BM, Draper GJ, Kingston JE. Retinoblastoma in Great Britain
1969-1980: incidence, treat ment, and survival. Br J Ophth 1988; 72: 576-583.
- Gunduz K, Shields Cl, Shields JA, Meadows AT. The outcome of
chemoreduction treatment in patients with Reese-Ellsworth group V
retinoblastoma. Arch Ophthalmol 1998; 116: 1.613-1.617.
- Yanoff m, Fine BS. Ocular Patthology. Lippincott Company 1994; 18: 693.
- He W, Hashimoto H, Tsuneyoshi MA. Reassesment of histologic clasification
and Immunohistochemical study of 88 retinoblastomas. Cancer 1992; 70:
2.901-2.908.
- Shields C, Shields JA, Baez K, Cater JR, De Potter P. Optic nerve
invasion of retinoblastoma. metástasis potential and clinical risk factors.
Cancer 1994; 73: 692-698.
- Khelfaoui F, Validire P, Auperin A, Quintana E. Histopathologic risk
factors in retinoblastoma. Cancer 1996; 77: 1.206-1.213.
- Apple DJ, Rabb MF. Retinoblastoma, leucocoria, and facomatosis. Ocular
Pathology. Clinical aplications and self assessment. Mosby Year Book Inc 1992;
8: 374-418.
- Shields JA, Shields CL, Eagle RC, Blair CJ. Spontaneous pseudohypopyon
secondary to diffuse infiltrating retinoblastoma. Arch Ophth 1998; 106:
1.301-1.302.
- Stafford WR, Yanoff M, Parnell B. Retinoblastoma initially misdiagnosed
as primary ocular inflammation. Arch Ophthalmol 1969; 82: 771-773.
- Shields CL, Shields JA, Shah P. Retinoblastoma in older children.
Opthalmology 1991; 98: 395-399.
- Abramson DH, Frank CM. Second nonocular tumors in survivors of bilateral
retinoblastoma; a possible age effect on radiation-related risk. Ophthalmology
1998; 105: 573-580.
- Roarty JD, McLean IW, Zimmerman LE. Incidence of second neoplasms in
patients with bilateral retinoblastoma. Ophthalmology 1988; 95: 1.583-1.587.
- Reese AB. Tumors of the eye. Hagertown, MD: Harper & Row 1976:
90-132.
- Abramson DH, Notterman RB, Ellswort, RM. Retinoblastoma treated in
infants in the first six months of life. Arch Ophthalmol 1986; 101: 1.362-1.366.
- Bagley LJ, Hurst RW, Zimmerman RA, Shields JA, Shields CL, De Potter P.
Imaging in the trilateral retinoblastoma syndrome. Neuroradiology 1996; 38:
166-170.
- Beets-Tan RGH, Hendricks M, Ramos LMP, TAN KEWP. Retinoblastoma: CT and
MRI. Neuroradiology 1994; 36: 59-62.
- Berrocal T, De Orbe A, Prieto C, Al-Assir I, Izquierdo C, Pastor I,
Abelairas J. US and Color Doppler Imaging of Ocular and Orbital Disease in the
Pediatric Age Group. Radiographics 1996; 16: 251-272.
- Berrocal T, De Orbe A, Prieto C, Bret M, Arjonilla A, Peralta J,
Abelairas J. Common and Uncommon US Manifestations of Retinoblastoma with
Pathologic Correlation. Ultrasound Med Biol 1997; 23: 81.
- Char DH, Miller TR, Ljung BM. Fine needle biopsy in retinoblastoma. Am J
Ophthalmol 1984; 97: 686-690.
- Shields JA, Shields CL, Suvarnamani C. Retinoblastoma manifesting as
orbital celulitis Am J Ophthalmol. 1991; 112: 442-449. Arch Ophthalmol. 1995;
113: 615-623.
- Kaufman LM, Mahmood F, Song D. Retinoblastoma and simulating lesions.Role
of CT, MR Imaging and use of Gd-DTPA contrast enhancement. Radiologic Clinics of
North America 1998; 36.6: 1.101-1.116.
- Liang JC, Augsburger JJ, Shields JA. Diffuse infiltrating retinoblastoma
associated with persistent primary vitreous. J Pediatr Ophthalmol Strabismus
1985; 22: 31-33.
- Messmer EP, Heinrich T, Hopping W. Risk factors for metastases in
patients with retinoblastoma. Ophthalmology 1991; 98: 136-141.
- Bhatnagar R, Vine AK. Diffuse infiltrating retinoblastoma. Ophthalmology
1991; 98: 1.657-1.661.
- Eng C, Lin FP, Abramsom DH. Mortality from second tumors among long-term
survivors of retinoblastoma. J Natl Cancer Inst 1993; 85: 1.121-1.128.
- Shields CL, Shields JA, Baez KA. Choroidal invasion of retinoblastoma:
metastatic potential and clinical rick factors Br J Ophthalmol 1993; 77:
544-548.
- Shields JA. Misconceptions and tecniques in the managment of
retinoblastoma. The 1992 Paul Hendkind Memorial Lecture. Retina 1992; 12:
320-330.
- Irvine AR, Albert DM, Sang DN. Retinal neoplasia and dysplasia.
Retinoblastoma ocurring with PHPV. Invest Ophthalmol Vis Sci 1967; 16: 403:407.
- Morgan KS, Mc Lean IV. Retinoblastoma and PHPV ocurring in the same
patient. Ophthalmology 1981; 88: 1.087-1.091.
- Offret H, Saraux H, Limon S. Dysgenesies vitreo-retiniennes. Persistance
et hyperplasie du vitre primitif. Leur traitement. Arch Ophthalmol (Paris) 1976;
36: 491-508.
- Bishop JO, Madsen EC. Retinoblastoma: Rewiew of current status. Surv
Ophthalmol 1995; 19: 342-366. Am J Ophthalmol 1979; 87: 276-277.
- Brown G, Shields JA, Oglesby RB. Anterior polar cataracts associated with
bilateral retinoblastoma.
- Friendly DS, Parks MM. Concurrence of hereditary congenital catarats and
hereditary retinoblastoma. Arch Ophthalmol 1970; 84: 525-527.
- Makley TA. Retinoblastoma in a 52 year old man. Arch Ophthalmol 1963; 69:
325.
- Shields JA. Retinoblastoma in an 18 year old male. Arch Ophthalmol 1976;
13: 274-277.
- Takahashi T. Retinoblastoma in a 26 year-old adult. Ophthalmology 1983;
90: 179-183.
- Kiratli HK, Bilgic S, Ozerdem U. Management of massive orbital
involvement of intraocular retinoblastoma. Ophthalmology 1998; 105: 322-326.
- Murphree AL, Villablanca JG, Deegan WF III, Sato JK, Malogolowkin M,
Fisher A, Parker R, Reed E, Gomer CJ. Chemotherapy plus local therapy in the
treatment of intraocular retinoblastoma. Arch Ophthalmol 1996; 114(11):
1348-1356.
- Cormack RA, Kooy HM, Bellerive MR, Loefller JS, Petersen ra, Tarbell NJ.
A stereotactic radiation therapy device for retinoblastoma using a noncircular
collimator and intensity filter. Med Phys 1998; 25: 1438-1442.
- Croughs P, Deman C, Richard F, Vynckier S, Van Obbergh L. Treatment of
retinoblastoma using accelerated protons. Bull Soc Belge Ophthalmol 1992; 243:
81-85.
- Doz F, Neusenschwander S, Plantaz D. Etoposide and carboplatin in
extraocular retinoblastomas: a study by the Societe Francaise d´Oncologie
Pediatrique. J Clin Oncol 1995; 13: 902-909.
- Gallie BL, Budning A, DeBoer G, Thiessen JJ, Koren G, Verjee Z, Ling V,
Chan HSL. Chemotherapy with Focal Therapy Can Cure Intraocular Retinoblastoma
without Radiotherapy. Arch Ophthalmol 1996; 114: 1.321-1.328.
- Journee-de Korver JG. Tranpupillary Thermotherapy: Tesis. The Hague, The
Netherlands: Kugler Publications; 1998; 1: 116-119.
- Roarty JD, McLean IW, Zimmerman LE. Incidence of second neoplasms in
patients with bilateral retinoblastoma. Ophthalmology 1988; 95: 1583-1587.
- Saarinen UM, Sariola H, Hovi L. Recurrent disseminated retinoblastoma
treated by high-dose chemotherapy, total body irradiation, and autologous bone
marrow rescue. Am J Pediatr Hematol Oncol 1991; 13: 315-319.
- Shields CL, Shields JA. Recent developments in the management of
retinoblastoma. J Pediatr Ophthalmol Strabismus1999; 36: 8-18.
- Howard GM, Ellsworth RM: Diferential diagnosis of retinoblastoma. A
statistical survey of 500 children. I. Relative frequency of the lesions which
simulating retinoblastoma. Am J Ophthalmol 1965; 60: 610-618.
- Hungerford JL, Toma NMG, Plowman PN, Kingston JE. External beam
radiotherapy for retinoblastoma: Br J Ophthalmol 1995; 79: 109-111.
- Imhof SM, Mourits MP, Hofman P. Quantification of orbital and midfacial
growth retardation after megavoltage external beam irradiation in children with
retinoblastoma. Ophthalmology 1996; 103: 263-268.
- Nelson SC, Friedman HS, Oakes WJ. Successful therapy for trilateral
retinoblastoma. Am J Ophthalmol 1992; 114: 23-29.
- Shields JA, Shields CL, Ehya H. Atypical retinal astrocytic hamartoma
diagnosed by fine needle biopsy. Ophthalmology. 1996; 103: 949-952.
- Toma NMG, Hungerford JL, Plowman PN. External beam radiotherapy for
retinoblastoma: Lens sparing technique. Br J Ophthalmol 1995; 79: 112-117.
- Gallie BL, Phillips RA, Ellsworth RM, Abramson DH. Significance of
retinoma and phthisis bulbi for retinoblastoma. Ophtalmology 1982 b; 89:
1.393-1.399.
- Eagle RC, Shields JA, Donoso L, Milner RS. Malignant transformation of
spontaneously regressed retinoblastoma, retinoma, retinocytoma variant.
Ophthalmology 1989; 96: 1.389-1.396.
- Sanborn GE, Augsburger JJ, Shields JA. Spontaneous regression of
bilateral retinoblastoma. British J Ophthalmol 1982; 66: 685-690.
- Abramson DH. Retinoma,retinocytoma, and retinoblastoma. Arch Ophthalmol
1983; 101: 1.517-1.518.
- Zimmerman LE. Retinoblastoma and retinocytoma. In Spencer WH, Font RL,
Green WR et al. Ophthalmic Pathology. An atlas and texbook, vol 2 ed 3. WB
Saunders Philadelphia 1985: 1.292-1.351.
- Lueder GT, Heon E, Gallie BL. Retinoma associated with vitreous seeding.
Am J Ophthalmol 1995; 119: 522-523.
- Gallie BL, Ellsworth RM, Abramson DH, Phillips RA. Retinoma:spontaneous
regression of retinoblastoma or benign manifestation of a mutation? Bri J Cancer
1982; 45: 513-521.
- Shields J, Parsons H, Shields C. Lesions simulating retinoblastoma. J
Pediatr Ophthalmol Strabismus 1991; 28: 338-340.
- Reese AB. Persistent hyperplastic primary vitreous. Am J Ophthalmol 1955;
40: 317-318.
- Balazs EA. Fine structure of the developing vitreous. Int Ophthalmol Clin
1973; 13(3): 169-187.
- Awan KJ. Thumayam M. Changes in the contralateral eye in uncomplicated
persistent hyperplastic primary vitreous. Am J Ophthalmol 1985; 99: 122-124.
- Jones H. Hyaloid remnants in the eyes of premature babies. Br J
Ophthalmol 1963; 47: 39-41.
- Renz B, Vygantas C. Hyaloid vascular remnants in human neonates. Ann
Ophthalmol 1977; 9: 179-184.
- Haddad R, Font RL, Reeser F. Persistent hyperplastic primary vitreous. A
clinicopathologic study of 62 cases and review of the literature. Surv
Ophthalmol 1978; 23: 123-134.
- A joint Statement of the American Academy of Pediatrics, the American
Association for Pediatrics Ophthalmology and Strabismus, and the American
Academy of Oftalmology. Screening Examination of premature infants for
Retinopathy of Prematurity. Ophthalmology 1997; 104: 888-889.
- Teske M, Trese M. Retinopathy of prematurity-like fundus and persistent
hyperplastic primary vitreous associated with maternal cocaine use. Am J
Ophthalmol 1987; 103: 719-720.
- Fonseca A, Peralta J, Abelairas J. Retinopatia del prematuro. Guía
práctica. Madrid 1996.
- Tasman WS, Augsburger JJ, ShieLds JA. Familiar exudative
vitreoretinopathy. Trans Am Ophthalmol Soc 1981; 79: 211-226.
- Shields JA. Ocular toxocariasis. A rewiew. Survey Ophthalmol 1984; 28:
361-381.
- Shield Shields JA, Lerner HA, Felberg NT. Aqueus cytology and enzymes in
nematode endophthalmitis. Am J Ophthalmol 1977; 84: 319-322.
- Shields JA, Felberg NT, Federman JL. Discussion of ELISA for the dignosis
of ocular toxocariasis. Ophthalmology 1979; 86: 750-752.
- Ridley ME, Shields DS, Brown GC, Tasman WS. Coats disease. Evaluation and
management. Ophthalmology 1982; 89: 1.381-1.387.
- Peralta J, Abelairas J, Fernández Guardiola JM, Fonseca A. Diagnóstico
del retinoblastoma: Importancia del calcio. Arch Soc Españ Oftalmol 1994; 67:
533-538.
- Richards WW. Retinoblastoma simulating uveitis. Am J Ophthalmol 1968; 65:
427.
- Shields JA, Parsons HM, Shields CL, Shah P. Lesions simulating
retinoblastoma. J Pediatr Ophthalmol Strabismus 1991; 28: 338-342.
- Shields JA, Shields CL, Parsons HM. Rewiew: differential diagnosis of
retinoblastoma. Retina 1991; 11: 232-243.
- Abelairas J, Perera A,Regules E, Gutiérrez M, Fernandez Guardiola JM,
Morales C, Romo A, Chacon J, Villarejo FJ, Perez Higueras A, Alfonso JM, Prieto
C, Pascual Castroviejo I. Meduloepitelioma (Dictioma ) del nervio optico. Arch
Soc Esp Oftal 1990; 58: 385-392.
- Palmer ML, Carney MD, Combs JL. Combined hamartomas of the retinal
pigment epithelium and retina. Retina 1990; 10: 3-36.
- Bouzas EA, Parry DM, Eldridge R, Kayser-Kupfer MI. Familiar occurrence of
combined pigment epithelial and retinal hamartomas associated with
neurofibromatosis 2. Retina 1992; 12: 103-107.
|





