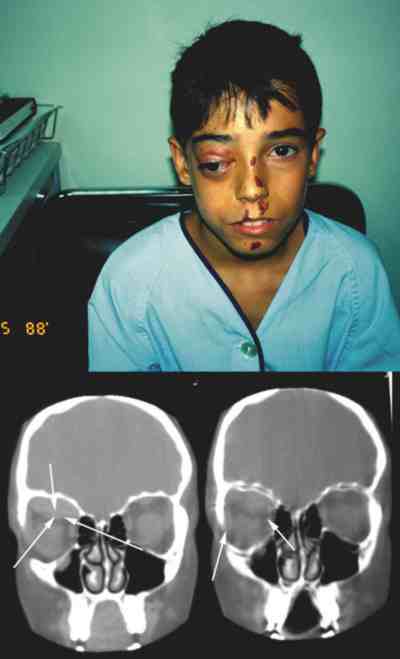
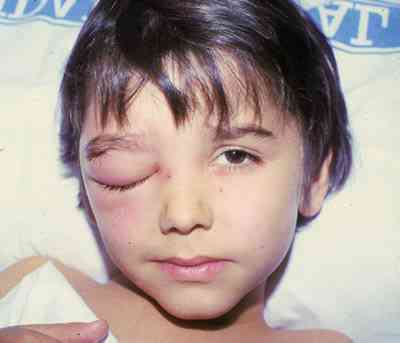
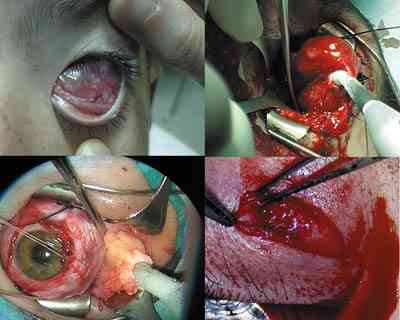
|
La aparición sistemática de series pediátricas dentro de las estadísticas de adultos hace bastante complejo el delimitar los campos de actuación de los diferentes especialistas que tratan la órbita y sus referencias son distintas dependiendo del enfoque clínico, quirúrgico y su integración en unidades craneofaciales y oculoplásticas. Son ya clásicas las aportaciones de Reese (1), Lloyd (2), Henderson (3,6), Jakobiec (4), Ira S. Jones (5), Rootman (7), Shields (8), Taylor (9) y en nuestro país Gutiérrez Díaz (10,11), Pérez Moreiras (12), Gil-Gibernau (13,15) y Zaragoza (14). Mención especial merece John Wright con mucho el cirujano de órbita más completo y con mayor experiencia en la historia, acumulada durante cuarenta años como centro de referencia en Londres. Igualmente Carlos García Alíx, pionero en España de la cirugía moderna orbitaria y un acérrimo defensor dentro de su idea global y práctica de la órbita en abordajes anteriores y laterales.Su visión multidisciplinaria y colaboración con otras especialidades en la Clínica Puerta de Hierro (Dres. Bravo, Carrillo, Garzón, Parera, Nombela y Brasa) en los dos primeros TAC que se introdujeron en España, hace 23 años, permitió familiarizarnos con la imágenes anatómicas que la tomografía axial computarizada aportaba en aquellos primeros prototipos de scanners (16,17). Su amistad con el Dr. Wright del Moorfields Hospital en Londres nos consiguió rotaciones y adquisición de experiencias en el mayor centro de referencia que existía sobre tumores en aquella época. Han pasado muchos años, con muertes, jubilaciones, cambio de especialidades y creación de unidades de órbita, oculoplástica y craneofacial. Sin embargo, en patología pediátrica, son los temas orbitarios que se relacionan con modernos métodos de diagnóstico y terapéutica, cada vez más interesantes. Por su variedad, las controversias y resultados interdisciplinarios se hacen más complejos y es difícil conseguir una visión integrada de los procesos.
CONSIDERACIONES GENERALES La incidencia de las diferentes proptosis y masas de afectación orbitaria en niños varía dependiendo de los estudios, centros de referencia, fuentes de la patología, países y continentes. Así las publicaciones de los Shields en Filadelfia (18) basadas en cirugías confirmadas con anatomía patológica, dada su especificidad como centro de referencia oncológico mostraban que las lesiones quísticas eran la causa más común de biopsias orbitarias. Al utilizar sólo las muestras quirúrgicas, los procesos infecciosos que son tan corrientes en la infancia y dan pseudoproptosis, no son contemplados y se hacen pocas tomas biópsicas al curarse con antibióticos. Igualmente hay patologías en procesos sistémicos con manifestación orbitaria (linfomas, leucemias), que se archivan por Patología en otras especialidades diferentes a Oftalmología y sólo se biopsian las zonas con mejor abordaje quirúrgico. Los trabajos de Rootman (7) y Bullock (19) que incluyen casos diagnosticados clínicamente, revelan que las patologías inflamatorias/infecciosas y los tumores vasculares y neurales eran más comunes de lo habitual. Paradójicas parecen en nuestro medio las series de Silva (20) en 1968 con 65 casos de tumores orbitarios infantiles por debajo de los 10 años de los cuales 20 correspondían a retinoblastomas con extensión orbitaria. Igualmente la de Belmekki (21) en 1999 tenía 13 retinoblastomas extra oculares de un total de 54 tumores pediátricos. Las variaciones geográficas también son llamativas. Templeton (22) trabajando sobre neoplasias de la infancia en el continente africano manifiesta que el linfoma de Burkitt es en su estadística la más significativa.
K. Sindhu y F. Martin (23) difieren en su artículo con Shields, Rootman y Bullock considerando tan solo 1 dermoide como causa de proptosis y excluyendo los 55 quistes que tenían con afectación orbitaria por considerar su incidencia en el desplazamiento ocular mínima. Por otra parte en los estudios de neuroimagen, los mucoceles y las sinusitis son la causa más frecuente de proptosis unilateral en Radiología (24,25). Como resumen se aporta que pese a las variaciones en las publicaciones, países, especialistas y centros de referencia (19,24,25).
Es muy importante para la cirugía de la órbita en edad pediátrica el dominio del complejo entramado anatómico, las relaciones con estructuras vecinas y el conocimiento de las manifestaciones clínicas de las lesiones orbitarias (26,27). La exploración en consulta de una órbita en la infancia, es muy diferente a la revisión oftalmológica general (28). La frecuencia y localización de la patología, unido al avance de las técnicas neurorradiológicas (TC helicoidal, RNM) han desarrollado y mejorado el diagnóstico y la actitud terapéutica (29).
INDICACIONES QUIRÚRGICAS (30-35) Lesiones encapsuladas Como regla general las masas encapsuladas como hemangiomas, neurinomas o quistes dermoides son más fácilmente extirpables. Sin embargo los coristomas, que frecuentemente aparecen en el cuadrante superoexterno de la órbita y se resecan con facilidad, pueden extenderse posteriormente hacia el ápex orbitario e incluso llegar hasta el compartimento cerebral.
Lesiones infiltrativas La excisión completa de tumores infiltrativos difusos es imposible de realizar y el riesgo de lesionar estructuras vitales es importante. Los pseudotumores responden bien a los corticoides y los rabdomiosarcomas se tratan con quimioterapia y radioterapia requiriendo una extirpación subtotal dentro de las posibilidades de cada cirujano. La cirugía sobre los gliomas del nervio óptico produce la pérdida de la visión que tengan, por lo que se suele esperar, pero hay que vigilarlos para que no lleguen al quiasma. La extirpación por vía neuroquirúrgica y el momento de realizarla es muy controvertida. Si recidivan en la órbita los retinoblastomas y rabdomiosarcomas tras consumir todas las medidas oncológicas y de radiación, nos obligan extirpaciones radicales previa confirmación histológica de recurrencia o remisión incompleta del tumor.
Lesiones en glándula lagrimal Afortunadamente en la infancia esta patología es poco frecuente pero es tan comprometida como en los adultos, con gran riesgo de diseminación y recurrencias. La mayoría de las veces son quistes o rabdomiosarcomas que comprimen la glándula y artefactan los estudios neurorradiológicos confundiéndolos con ella. Las inflamaciones glandulares son infiltrativas y se adaptan a las paredes orbitarias contorneando el globo ocular. Los pseudotumores y leucemias de diverso grado que se localizan en el cuadrante superoexterno de la órbita siguen sin tipificarse claramente, son más sólidos y producen mayor desplazamiento. Muchas veces los acinus son desestructurados por estas lesiones con fibrosis intralacrimal. Por otra parte las neoplasias epiteliales en la infancia son escasas y se manifiestan principalmente a partir de los 10 años. Los tumores mixtos benignos y los adenoquistocarcinomas son mucho más infrecuentes. Las parestesias por extensión perineural del tumor, el dolor y el crecimiento rápido ensombrecen el pronóstico. El dilema que en los adultos se plantea en cuanto a conservar la cápsula, programar las biopsias incisionales e intraoperatoras y extirpar en bloque con márgenes de seguridad todo reborde orbitario, se traslada igualmente a la infancia. Es fundamental diagnosticar el tumor involucrando en el mismo acto quirúrgico a los diferentes especialistas para conseguir buenas supervivencias. Resaltar la necesidad de hacer exhaustivos controles sistémicos oncológicos con técnicas de imagen. Hay que consultar con el anatomopatólogo el método óptimo para manejar el tejido extirpado. Actualmente se necesita extraer bastante cantidad de muestras en fresco para estudios de citología immunohistoquímica y genética.
Lesiones residuales Admiten múltiples variedades dependiendo del tipo de tumor, tanto por ciento de extirpación realizada, tratamiento con quimio y/o radioterapia, cicatrización y fibrosis en áreas con desestructuración quirúrgica. Tenemos que valorar exhaustivamente las exploraciones complementarias para estar seguros de la indicación operatoria y trasladar a la familia la necesidad de realizar operaciones mutilantes en pacientes que han consumido todas las posibilidades terapéuticas. Por suerte en la literatura (35) cada vez son más numerosos los grupos interdisciplinarios que trabajan en unidades craneofaciales y la publicación de series y posibilidades terapéuticas ordenadas y con gran credibilidad, ajenas al lucimiento personal, son de gran ayuda para los que nos dedicamos a esta patología.
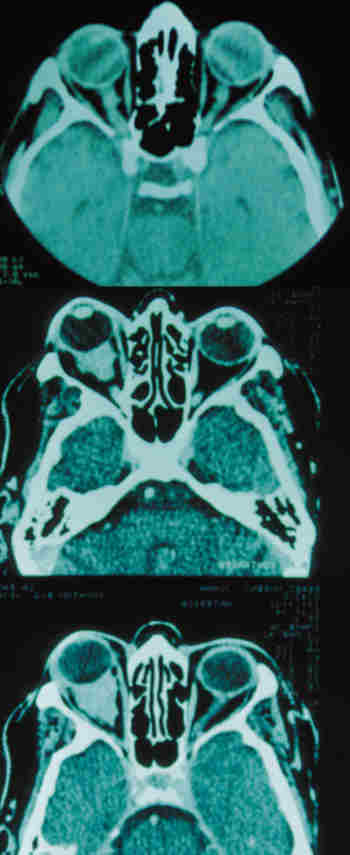
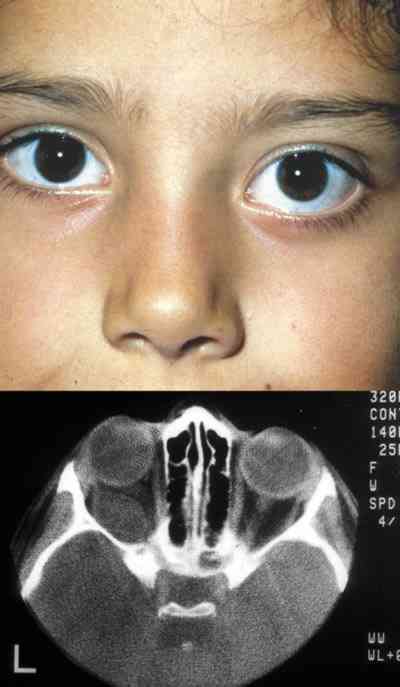
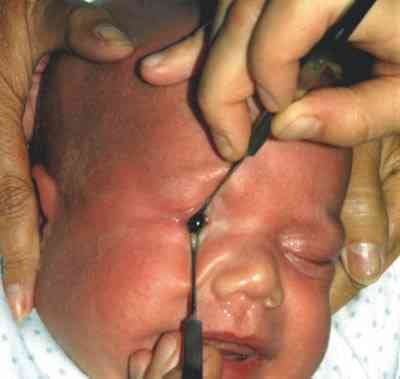
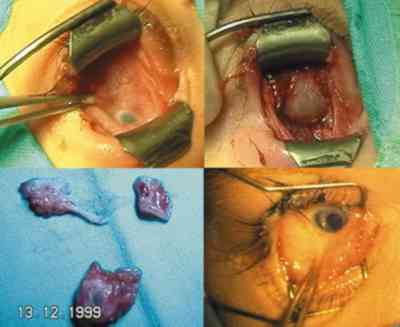
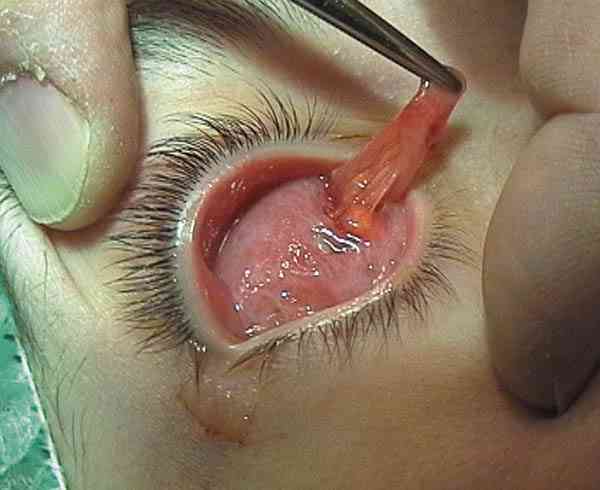
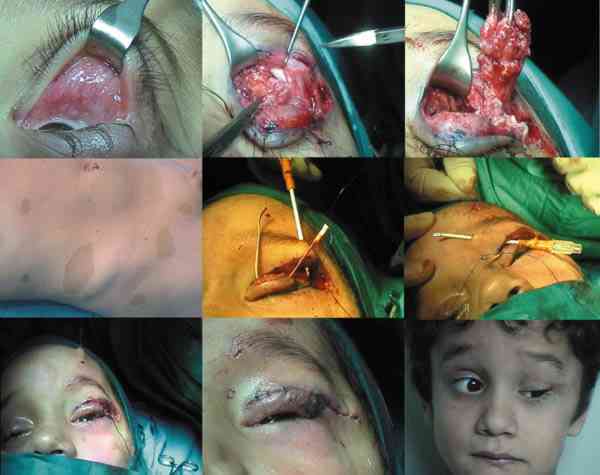
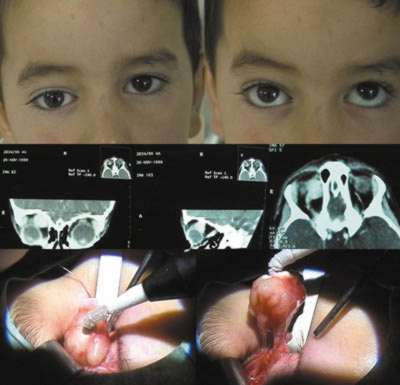
QUISTES, ANOMALÍAS CONGÉNITAS Y ECTOPIAS Coristomas (dermoides y otros quistes epiteliales) Son más frecuentes en los cuadrantes superotemporales de la órbita. Durante la embriogénesis acúmulos de células epiteliales quedan atrapados cerca de las uniones frontozigomáticas y adoptan apariencias quísticas. En vez de dermoide el término coristoma con proliferación de los componentes dérmicos los define mejor. Contienen queratina, pelos y en sus cubiertas epiteliales encontramos anexas glándulas sebáceas, folículos pilosos, glándulas sudoríparas y lacrimales (36).
Si las cápsulas no son tan complejas se llaman epidermoides. Si hay mayor proporción de tejido adiposo dermolipomas. En los quistes intraorbitarios puede aparecer epitelio conjuntival e incluso del tracto respiratorio. Pueden localizarse al momento del nacimiento o aparecer más tarde con el desarrollo. Los quistes ductales del sistema lacrimal suelen aparecer en la parte palpebral y glándulas accesorias. En la región orbitaria son más escasos y de mayor complejidad (37).
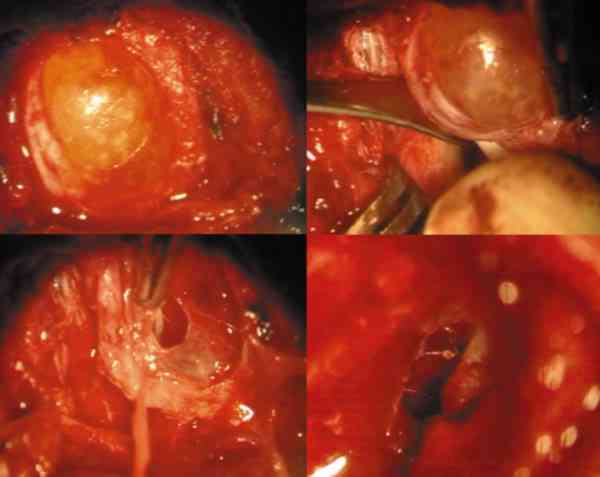
La ruptura por traumatismos o maniobras quirúrgicas, producen una gran reacción inflamatoria y granulomatosa en la órbita por lo que su resección in toto, con disección completa de la cápsula es obligatoria. La marsupialización de las masas cuando son muy grandes permite su extracción posterior con más facilidad y la tenemos que emplear frecuentemente (38-41).
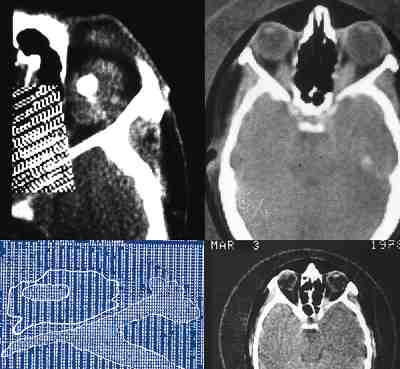
Craneosinostosis Se producen por cierre de las suturas prematuramente produciendo proptosis del contenido orbitario al crecer y antero posición del ala mayor del esfenoides. Síndromes como el Crouzon, Apert etc., dan manifestaciones oftalmológicas urgentes como luxaciones oculares, papiledema y atrofias ópticas que precisan descompresiones neuroquirúrgicas urgentes y centralización de los pacientes dentro unidades craneofaciales (42-46).
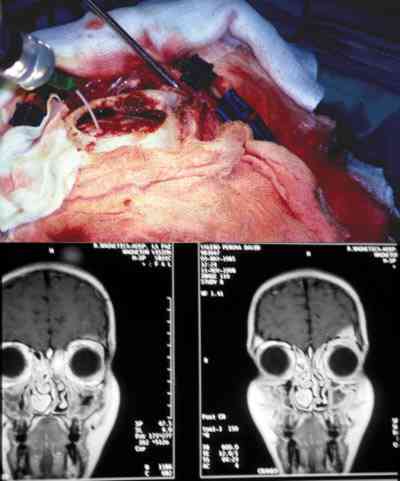
Cefaloceles Presencia de tejido glial en la órbita en anomalías del desarrollo y malformaciones. Desplazan al globo ocular, siendo la proptosis pulsátil y según su composición la herniación de los tejidos cerebrales adopta los nombres de encefalocele, meningocele, meningoencefalocele, etc. (47). Otras veces los quistes gliales parten de las cubiertas del nervio óptico o se combinan con colobomas y microftalmías que obligan a reconstrucciones de cavidades anoftálmicas complejas. La aparición de fístulas de líquido cefalorraquídeo exige actuaciones combinadas con los neurocirujanos (47-49). El control con técnicas de imagen resulta fundamental para delimitar campos, evolución y actuaciones quirúrgicas.
Microftalmos o anoftalmos con quiste Nunca hay un verdadero anoftalmos. En nuestra experiencia siempre existe un rudimento minúsculo que puede identificarse como un esbozo ocular. Pueden ser uni o bilaterales, asociados con colobomas y malformaciones cerebrales (50-52). A veces el quiste persiste y crece lentamente durante años, permitiendo un desarrollo armónico de la cavidad orbitaria. Si se dilata mucho tenemos que extirparlo y ello conlleva sacrificar el ojo microftálmico colocándose un implante integrado (52-55). Si hay comunicación con el líquido cefalorraquídeo por algún componente glial y se rellena la órbita, tenemos que localizar la fístula usando incluso abordajes craneales (56).
Glándulas lacrimales ectópicas En párpados, conjuntiva, cornea, coroides e iris. Dan proptosis y su exéresis si no es total pueden dar tumores malignos como adenocarcinomas y adenomas pleomorfos (57-58).
Quistes de inclusión y conjuntivales Son congénitos o secundarios a traumatismos o cirugías. Si no se extirpan en su totalidad, recurren al quedar restos de cápsulas. Son muy aparatosos y son remitidos por oftalmólogos a las unidades infantiles con múltiples y pintorescos diagnósticos (59-62).
Quistes hidatídicos Infestación secundaria por parásitos intestinales (equinococus). Tienen calcio en la cápsula y requieren su extirpación total, por el peligro de las recidivas. La eosinofilia y el ELISA son patognomónicos. La cisticercosis y triquinosis pueden dar también afectación quística orbitaria (63-65).
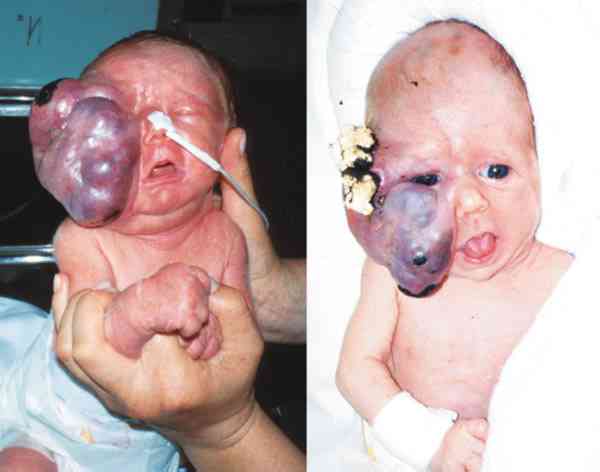
Teratomas Se desarrollan a partir de las 3 capas germinales, aunque en algunos solo hay 2 de ellas, siendo el mesodermo el que está siempre presente. A veces tiene componentes dérmicos, del tracto respiratorio, intestinal y glandulares. Se ha dado el caso de fetos pobremente diferenciados en la órbita.
Se da en niñas con una frecuencia de 2:1 y en recién nacidos. La proptosis y el desplazamiento ocular son notorios (66-70).
Pueden estar limitados al compartimento orbitario o acceder en su extensión a la cavidad craneal y los senos paranasales (69-71). Igualmente son primarios o secundarios a partir de estructuras vecinas. Adoptan formas quísticas y fluctuantes. Su crecimiento es más agresivo y la extirpación es compleja precisando la colaboración de la unidad craneofacial (70-72). Los orbitarios raramente malignizan, aunque las recurrencias sí lo pueden hacer (72). Resecar quirúrgicamente el teratoma sin compromiso del globo ocular a veces es imposible (71-73).
Hamartomas Son crecimientos anómalos de tejido, compuesto por células maduras que se desarrollan de una manera desproporcionada en el área orbitaria natural donde habitualmente están presentes. Los angiomas capilares son los ejemplos más representativos (74-75).
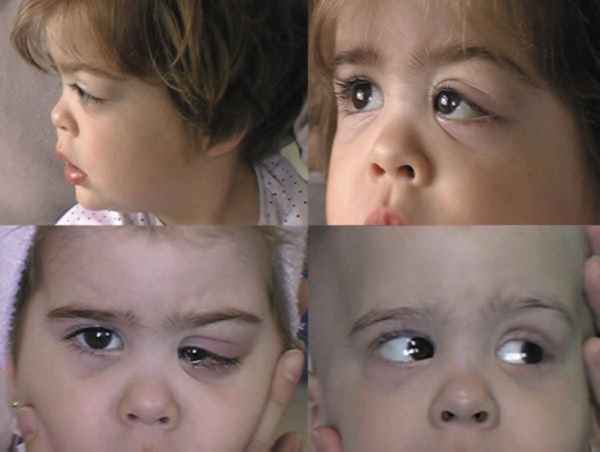
TUMORES MESENQUIMALES VASCULARES Angiomas capilares Es el más frecuente de los tumores vasculares en la infancia. Puede dar proptosis en las primeras 2 semanas de vida del niño. Se puede asociar con hemangiomas cutáneos y perioculares. Se exacerba con lloros y al colocarlos boca abajo. Crece durante meses. Su regresión con corticoides es espectacular aunque a veces si no se tratan disminuyen muy lentamente. El peligro de comprometer la agudeza visual, marca el límite intervencionista (75-78).
El Eco-Doppler color, la TC y la RNM los definen con gran flujo y vascularización (76). Bajo control pediátrico se acelera su regresión con esteroides sistémicos en pautas de varios meses. Si no remiten el interferón es la última modalidad terapéutica (79-80). Los corticoides intralesionales (betametasona y triamcinolona) producen reducciones rápidas pero también están descritas múltiples complicaciones ya que la embolización fortuita de los vasos intratumorales por vía retrógrada causa ceguera. La ecografía permite un correcto abordaje de la lesión. El TC helicoidal da mejor definición y es muy rápido (77-80).
La embolización en sospechas de malformaciones arteriovenosas, el uso de las sustancias esclerosantes y la excisión quirúrgica está muy contestada en la actualidad (76). Sólo en casos especiales de fácil abordaje y cuando el tumor manifiesta su regresión se podría intentar esta última técnica (77). La radioterapia causa demasiados problemas secundarios y está contraindicada. Su uso en casos dramáticos refractarios a toda terapia, da complicaciones secundarias y posibilidad de osteosarcomas por radiación (81-82). Los modernos láseres estéticos no permiten la penetración en toda la masa tumoral y sólo en regresiones y resecciones cutáneas tiene su indicación plástica (78). Durante la evolución se producen astigmatismos muy marcados por lo que las correcciones ópticas y terapias oclusivas son fundamentales.
Angiomas cavernosos Es la causa más frecuente de proptosis en el adulto, mientras que son muy raros en la edad pediátrica. Son lesiones intracónicas y de localización en los cuadrantes inferolaterales. Las técnicas de neuroimagen moderna los diagnostican sin dificultad (82-83). A veces si son muy posteriores es preciso realizar orbitotomías laterales y transcraneales (83-84).
Hemangiopericitoma Raros, encapsulados, similares a los angiomas cavernosos del adulto pero más complejos. Crecimiento más rápido con quemosis conjuntival y muy infrecuentes en la infancia. El diagnóstico suele ser por anatomía patológica y diferido (85-88). La clínica y métodos de diagnóstico radiológicos nunca se aproximan.
Su extirpación referida en la literatura debe ser total, porque puede recurrir, malignizarse y dar metástasis, aunque con grandes intervalos de tiempo (87,88). Sullivan refiere 5 características para diferenciarlo y llegar a una presunción siendo la última una arteriografía carotídea. En su serie (89) de 12 tumores en 23 años extraídos del Moorfields Eye Hospital reconoce la dificultad en la exéresis completa y sobre todo su diagnóstico preoperatorio.
SÍNDROMES DE ENMASCARAMIENTO Fístulas arteriovenosas Conexiones entre venas y arterias que previamente a un trauma eran normales. En la infancia y en la literatura son muy raras. Las carótido cavernosas dan las típicas cabezas de medusa epibulbares y epiesclerales, con proptosis pulsátil, ruido, glaucoma y parálisis del sexto par. Las dúrales son más insidiosas y producen ojo rojo crónico. En las técnicas de imagen se observa dilatación de la vena oftálmica superior por arteriolización y engrosamiento difuso de toda la musculatura orbitaria. Arteriografías selectivas y embolizaciones son técnicas muy sofisticadas que precisan gran especialización en el tema y muchas veces los resultados son sólo paliativos (8,75,82). El síndrome de Wiburn-Mason, consistente en aneurismas congénitos arteriovenosos de la retina y cerebro a veces tiene afectación orbitaria ipsilateral (90).
Malformaciones vasculares/linfangioma Son malformaciones vasculares con presencia de componentes venosos y linfáticos. Muy infrecuentes, se presentan en el primer quinquenio de la vida. Se localizan en la conjuntiva, párpados, órbita, orofaringe y senos paranasales (82). Pueden manifestar proptosis al nacimiento. Tras catarros o traumatismos se exacerban debido a una probable respuesta inmunológica dentro del tejido linfoide de la lesión. Lentos en su crecimiento, casi nunca regresan. La afectación subcutánea hace que el párpado adquiera una coloración azul.
Cuando sangran se forman en los canales linfáticos ectásicos los quistes de chocolate y en la conjuntiva se aprecian dilatados y hemorrágicos (91-92). El Eco-Dopler color, el TC y la RNM revelan masas no encapsuladas, multiquísticas, irregulares de morfología anular con presencia de sangre intratumoral y bastante captación de contraste (76). El tratamiento quirúrgico es complicado porque nunca se consigue una extirpación completa y se comprometen estructuras palpebrales y fondos de saco que producen retracciones y hay que emplear cirugías reconstructoras complejas. Al no estar encapsulados y ser infiltrativas si nos introducimos en la órbita se pueden extirpar estructuras normales por no disponer de planos de clivaje para diseccionarlas. Excisiones parciales y drenaje de quistes hemorrágicos cuando son muy llamativos, solucionan temporalmente los cuadros clínicos y con la edad los brotes disminuyen (93). J.E. Wright ha publicado (94) que los linfangiomas orbitarios pertenecen al complicado campo de las anomalías venosas que se interrelacionan, e incluso parecen tener una solución de continuidad según su experiencia. Ya con el paciente en la edad adulta tuvo que extirpar y realizar orbitotomías descompresivas con abordajes laterales y transcraneales neuroquirúrgicos, por el gran compromiso para el globo ocular y el nervio óptico que los macroquistes producían. Se recomienda buscar malformaciones vasculares cerebrales que se asocian con linfangiomas en un 25% (92).
Malformaciones arteriovenosas Son anomalías del desarrollo compuestas de anastómosis anómalas entre arterias y venas sin lecho capilar que las interrelacione (94-97). A veces se asocian con afectación cerebral y su tratamiento quirúrgico pasa por un buen estudio angiográfico con oclusión selectiva de los vasos aferentes y excisión de la malformación (76,96-98). La radiocirugía con Gamma Knife o LINAC y las modernas técnicas de implantes radiactivos intralesionales tienen futuro pero en la órbita por ahora no están desarrolladas y en el cerebro puede haber recurrencias y complicaciones (96-101).
Varices Dilatación de las venas existentes en zonas orbitarias. Se manifiestan en edad juvenil-adulta. Pueden dar hemorragias y trombosis con lo que la proptosis intermitente que ordinariamente solo se observa en situación de pronación o con la maniobra de valsalva se convierte en fija. En casos raros se pueden asociar con varices intracraneales (94,102-103). El Eco-Doppler color y el TC helicoidal hechos en decúbito prono, con valsalva o presión sobre la yugular son diagnósticos. A veces se observan flebolitos (76). El tratamiento es conservador porque los hematomas desaparecen con el tiempo y es imposible una completa resección quirúrgica. Los riesgos de afectación visual y lesión de estructuras nobles en la órbita si les operamos son muy grandes (102,103). La embolización con partículas (104) por vía venosa retrógrada está referida en la literatura igualmente como paliativa con mejorías parciales. La cirugía con extirpación, o con disección y embolización in situ también está descrita, pero en las profundas implica un alto riesgo para la visión que hay que valorar (102). En la zona del saco lagrimal hay que hacer diagnóstico diferencial con mucoceles etmoidales por su similar apariencia clínica (105).
Hemorragias orbitarias Secundarias a trauma, rotura de malformaciones arteriovenosas, linfangioma o esfuerzos violentos (102). Están descritas hemorragias al practicar la maniobra de valsalva (106). La RNM en series sucesivas de reabsorción del hematoma permite observar la evolución sin ser intervencionista (76). El drenaje y la aspiración sólo es necesario si la visión está comprometida. La compresión del nervio óptico y el aumento de la presión orbitaria impide la perfusión arterial del ojo. Son más frecuentes los quistes hemáticos subperiósticos que tardan bastante en reabsorberse y que tenemos que evacuar por vía transcutánea anterior si hay problemas (107-108).
INFLAMACIONES La patología inflamatoria de la órbita adopta en su estructura diferentes localizaciones y dentro de la infancia las infecciones son las más frecuentes.
Infecciosas El considerar si la celulitis es preseptal u orbitaria, en disquisiciones sin importancia artefacta los diagnósticos y retrasa los tratamientos, porque los espacios están interrelacionados y el inicio de un orzuelo en condiciones de inmunosupresión puede extenderse a otros compartimentos, desarrollar abscesos subperiósticos, osteomielítis y afectación cerebral en diversas manifestaciones y formas. Aun así el septum orbitario delimita la gravedad del proceso y precisa cuando obligatoriamente debemos ingresar al niño en el hospital (109-112).
Una sinustis etmoidal o un flemón dentario pueden continuarse con una inflamación intraconal que afecta a la motilidad ocular, con un absceso y extensión al ápex orbitario que compromete a la visión, e incluso llega al seno cavernoso produciendo una trombosis que hace peligrar la vida del paciente (112-115). Si atraviesan las hendiduras y agujeros óseos alcanzan las meninges y el cerebro dando inflamaciones y abscesos craneales (112-114). El ingreso con estudio microbiológico, cobertura antibiótica apropiada y exploraciones neuroradiológicas sensatas es obligatorio (112-114). Hay que hacer tratamientos sintomáticos y de vecindad, teniendo las ideas claras. Si evolucionan a abscesos hay que valorar cuándo deben ser drenados quirúrgicamente. Es preceptivo utilizar las vías de abordaje correctas con estudios de neuroimagen que descarten claramente la afectación de espacios limítrofes (114,115).
Infecciones bacterianas. Contigüidad de sinusitis, trauma, cuerpos extraños y bacteriemia representan por este orden su incidencia etiológica. Si realizamos cultivos y antibiogramas de las muestras y cavidades afectadas sistemáticamente el Hemofilus influenza, estreptococo y estafilococo áureo son los organismos más frecuentemente encontrados (115-118). García y Harris proponen un tratamiento conservador para los abscesos orbitarios en niños menores de 9 años. Consiguieron evitar en un 93% la cirugía de evacuación y el drenaje subperióstico (117).
No infecciosas Ya hemos explicado en otro capítulo que la orbitopatía tiroidea es muy escasa en la infancia Tan sólo afectaciones palpebrales y mínimas infiltraciones musculares se reflejan en la literatura (119,120).
Los pseudotumores, granulomas, miositis y adenitis al ser polimorfa su composición inflamatoria admiten megadosis de corticoides y su respuesta terapéutica es bastante eficaz. Con el tiempo la fibrosis reemplaza a la grasa orbitaria y encapsula los músculos y el nervio óptico con lo que no hay respuesta (121,123).
PSEUDOTUMOR ORBITARIO Enfermedad inflamatoria idiopática de la órbita. Etiología desconocida. Produce proptosis y/o desplazamiento del globo. Comienzo agudo o subagudo, con dolor, restricción de movimientos, quémosis conjuntival y edema palpebral. Un tercio de los casos son bilaterales. Son el 8,5% de las patologías orbitarias de presentación tumoral. El 7% de los pseudotumores son pediátricos (123-124). Las técnicas de imagen son variopintas en su diagnóstico. Varían en cada caso. Las masas pueden ser irregulares o bien definidas con afectación única o de varios tejidos orbitarios. Hay que descartar y establecer asociaciones con enfermedades autoinmunes mediante estudios pediátricos completos que van más allá de las atribuciones de un oftalmólogo. Por eso la importancia de especialistas y oncoterapeutas (114). Las miositis de uno o más músculos, compromiso de las vainas del nervio óptico, de la esclera, coroides o de todos los tejidos son frecuentes. A veces su presentación es atípica, sin dolor y con mínima inflamación (119,120). Si se tratan con corticoides y antiinflamatorios orales responden bien en etapas precoces. El peligro es enmascarar una metástasis (linfoma) que responde espectacularmente y luego al rebrotar pasa a un estadio más avanzado. Los pediatras y oncólogos aconsejan no usarlos pero cuando hay compromiso de la agudeza visual nos vemos obligados a darlos intravenosos. Biopsias urgentes nos facilitan su utilización (122,123). Hay que mantener la terapia corticoidea durante meses, por el peligro de recurrencias. La anatomía patológica resalta inflamación pleomorfa y reacción tisular fibrovascular. La celularidad en su mayoría son linfocitos. A veces hay eosinófilos. No hay las células gigantes ni epitelioides típicas de las granulomato sis (122). Los oncoterapeutas son reacios a la utilización de corticoides por el peligro que los linfomas plantean en su diagnóstico diferencial. Éstos son más silentes, con poco compromiso visual. El problema es que la batería de pruebas diagnósticas que realizan e incluso la confirmación histológica demora en varios días el tratamiento y la fibrosis sustituye al componente inflamatorio por lo que la respuesta terapéutica es menor (122-125). La radioterapia puede ser la última posibilidad e incluso no responden a ella, quedando en la órbita masas densas que al resecarse quirúrgicamente están necróticas.
GRANULOMATOSIS DE WEGENER Patología multisistémica de etiología desconocida caracterizada como una vasculitis necrotizante granulomatosa (126-128). Su rareza en la infancia, con ocupación a veces de ambas órbitas, dolor, proptosis y afectación escleral es realmente dramática (127). La respuesta a corticoides no era demasiado buena antes de la combinación con antimitóticos (ciclofosfamida). También se utiliza la ciclosporina para las recurrencias. Puede infiltrar la glándula lacrimal y los músculos extraoculares. La cirugía sobre los granulomas no parece tener mucho éxito, utilizándose sólo como descompresión (129,130). Las biopsias y PAAF son complicadas. Para hallar el diagnóstico de vasculítis y hay que combinarlas con múltiples pruebas de laboratorio y microbiología (127,131,132). El tratamiento con trimetropin–sulfametoxazol se empleaba como primera elección, pero no parece ser efectivo en la actualidad. La radioterapia también es controvertida.
TUMORES ORBITARIOS MESENQUIMALES MUSCULARES Rabdomioma Hatsukawa presenta un niño de 16 meses con proptosis de 1 mes de evolución y resección parcial quirúrgica. La histología demostró las características benignas de la tumoración y durante 1,5 años de seguimiento no recibió tratamiento adicional, sin mostrar signos de recidiva (133).
Rabdomiosarcoma Es la más frecuente de la tumoraciones orbitarias en la infancia. La media de edad en su aparición son los 7-8 años. A veces es congénito. Resaltar que en su origen provienen de células precursoras pluripotenciales que luego se diferencian en músculo estriado. Así pues no salen de los músculos extraoculares ni tienen solución de contigüidad con ellos. Pueden rodearlos y establecer adherencias que resultan extremadamente difíciles de disecar. Es raro en la edad adulta. Puede metastatizar. No suele afectar a las paredes óseas. Actualmente los porcentajes de supervivencia sobrepasan el 95% si el tumor está confinado en la órbita (134-139).
Proptosis y desplazamiento ocular son de rápida aparición. Traumatismos accidentales o gradual crecimiento durante 1 mes, pueden retrasar el diagnóstico al no ser tan aparentes. Edema palpebral y quemosis lo confunden con pseudotumor orbitario o con alergias (140). Eco Doppler color, TC y RNM son obligatorios y definen masas irregulares u ovoideas, inicialmente bien circunscritas y posteriormente difusas, de localización en tejidos blandos orbitarios, que captan contraste (141,142). La biopsia intraoperatoria permite establecer el diagnóstico. Se necesita bastante material para congelación, microscopia óptica, electrónica e inmunohistoquímica. La cirugía pocas veces reseca todo el tumor ya que es friable y casi nunca pequeño y circunscrito. La extracción con crioterapia, sin dañar zonas vitales y ser meticuloso es fundamental. Saber concluir la cirugía si las adherencias e infiltraciones hacen peligrar la estructura orbitaria es de sentido común pero perjudica el ego de los cirujanos que requieren resultados espectaculares (135,140,143). Posteriormente la clasificación e inmunohistoquímica definen el tipo de tratamiento. Hay disparidad de criterios entre la escuela Europea y la Americana (134-136). En nuestro continente se somete a ciclos de quimioterapia que hacen desaparecer la tumoración o la reducen significativamente. Trasforman en masas menos friables y encapsuladas los restos del rabdomiosarcoma. De esta forma el abordaje quirúrgico posterior permite extirpar los restos y la anatomía patológica confirma un buen porcentaje de curaciones. Se evita los efectos secundarios de la radioterapia y si es necesario utilizarla los niños son como mínimo 1 año mayores con lo que resisten mejor la radiación (144).
En Estados Unidos, se combinan las dos técnicas como primera terapia. Son necesarias evaluaciones sistémicas por los onco y radioterapeutas ya que hay posibilidad de metástasis tanto regionales como a distancia. TC de tórax, biopsia y aspiración de médula ósea en combinación con la punción lumbar son obligatorias (135,136). Técnicas de futuro incluyen la radioterapia con control estereotáxico usando Gamma Nyfe y TC helicoidal. Igualmente el LINAC y el Acelerador de Protones están desarrollando protocolos para su uso en la edad pediátrica. Abramson ha publicado (145) la utilización de implantes radiactivos dentro de la órbita en recidivas pero la focalización de nuevas técnicas radioterápicas está todavía en desarrollo para la infancia.
TUMORES MESENQUIMALES FIBROHISTOCITARIOS Histiocitoma fibroso Son tumores frecuentes en adultos y muy raros en la infancia pero los hay descritos en niños de 6 meses (146,147). La localización es en la órbita nasal superior. La histología revela una mezcla de histiocitos, fibroblastos y células mesenquimales indiferenciadas. Aunque la mayoría son benignos hay variedades malignas. Cuando aparecen mitosis, pleomorfismo y necrosis los consideran malignos (148-150). La extirpación quirúrgica, nuevas cirugías y quimioterapia son los procedimientos de elección. La radioterapia adicional se emplea en la edad adulta (151,152).
Tumor fibroso solitario de la órbita Sólo había, en 1996, 8 casos descritos en la literatura. Proptosis de aparición gradual. Benigno, se extirpa con facilidad. Puede recurrir. Es un diagnóstico de anatomía patológica (153-156).
Fibromas Son rarísimos en la órbita y se presentan en la adolescencia. Fibroblastos y fibras de colágeno bien diferenciado son sus componentes. La cirugía extirpa fácilmente los de localización anterior y subconjuntivales. Los localizados en el saco y glándula lagrimal en sus disecciones son más complejos al estar adheridos a las paredes óseas, con fibrosis en las estructuras subyacentes. Nunca malignizan pero pueden recurrir. Los de cavidad nasal y áreas limítrofes de afectación orbitarias requieren colaboración con otras especialidades (18,156).
Fibromatosis Es una lesión con proliferación de fibroblastos, benigna pero localmente agresiva. Clínicamente y por anatomía patológica es una lesión intermedia entre la fibrosis y el fibrosarcoma. Puede ser solitaria en un 73%, multicéntrica (tejido subcutáneo, músculo esquelético, huesos largos) y generalizada con afectación visceral. Más corriente en niños en 2:1 y crece rápidamente en la órbita por lo que se confunde con fibrosarcoma. El tratamiento es quirúrgico y suele haber recurrencias (148,157,158).
Fascitis nodular Es una lesión fibrosa que afecta a párpados, conjuntiva, cápsula de tenon y órbita. Puede ser solitaria, nodular o infiltrar los músculos vecinos. Crece rápidamente, pareciendo una lesión maligna. La histología confirma su benignidad. Apariencia inflamatoria, con erosión focal del hueso adyacente (159,160).
TUMORES ÓSEOS Displasia fibrosa Puede aparecer en la órbita o ser poliostótica. El síndrome de Albright los asocia con hiperpigmentación cutánea y trastornos endocrinos. Se presenta en las primeras tres décadas de la vida. Como consecuencia de alteraciones en la maduración el hueso se desestructura, con estroma fibroso, focos líticos y áreas muy vascularizadas (161,162). El tumor cruza las líneas de sutura y afecta a múltiples huesos de la órbita. Se asocian con sarcomas óseos para complicar las patologías (163). Puede ocurrir estrechamiento del canal óptico y del sistema de drenaje lagrimal. Hay que practicar cirugía descompresiva en la mayoría por vía neuroquirúrgica, aunque afortunadamente se puede demorar hasta la edad juvenil sin compromiso de estructuras vitales (163-166).
Fibroma osificante juvenil Es una variante de displasia fibrosa benigna con características histológicas típicas y muchas espículas óseas con un estroma muy vascularizado. Los localizados en el ápex orbitario dan alteraciones del nervio óptico y defectos campimétricos. En el etmoides producen epífora por obstrucción lagrimal (167,168).
Es monostótica y no cruza las líneas de sutura. En los senos paranasales se complican con mucoceles (169,170). Requiere grupos multidisciplinarios para su cirugía con vías de abordaje consensuadas (171,172).
Osteomas Tumores óseos benignos que aparecen casualmente en los bordes del seno frontal o glabela sin causar problemas en su resección quirúrgica. Crecimiento lento, bien circunscrito y composición de hueso maduro. No está claro si los osteomas proceden de etiologías traumáticas, anomalías del desarrollo o infecciones. Mayor afectación en varones y no suelen causar síntomas antes de los 18 años. En el síndrome de Gardner (poliposis familiar) se asocian múltiples osteomas (172). La cirugía con extirpación del tumor se realiza cuando dan sintomatología y afortunadamente son raras en pediatría. Las operaciones multidisciplinarias con Neurocirujanos, Maxilofaciales y ORL se realizan cuando son adultos y hay compromiso del nervio óptico en las lesiones del esfenoides (173,174).
Quistes aneurismáticos Son masas benignas pseudotumorales de etiología desconocida. Parecen lesiones reactivas a traumatismos, afectaciones óseas, vasculares, y fibromas no osificantes de diversas localizaciones periorbitarias. Se tratan con exéresis locales o curetajes en combinación a veces con otras especialidades quirúrgicas. Se asocian más frecuentemente con displasias fibrosas. Los hay descritos desde los 14 meses de edad (175-177). Erosión de las paredes orbitarias y proptosis son sus características. Suelen dar manifestaciones clínicas en la adolescencia. Los espacios quísticos están rellenos de septos que contienen fibroblastos, histiocitos, osteoclastos y trabéculas de hueso rellenas de osteoblastos. Hay calcificaciones intalesionales (178,179). La radioterapia está contraindicada en la actualidad. La embolizacion selectiva de las lesiones favorece su calcificación (180-181).
Fibrosarcoma, condrosarcoma, osteosarcoma Tumoraciones que tienen en común la mayoría de las veces un glioma o un retinoblastoma radiado previamente con predisposición genética, apareciendo como segundas neoplasias varios años después.
El fibrosarcoma congénito y el infantil son raros, de crecimiento rápido y baja incidencia en metástasis. Los juveniles si se extirpan radicalmente tienen buen pronóstico (18,146,182).
Tumor de Ewing Representa el estadio final e indiferenciado de un espectro de células tumorales óseas de origen neural. La célula original todavía es controvertida. El tumor engloba huesos largos, costillas y pelvis. Afecta la órbita secundariamente con metástasis. El 75% de los pacientes son jóvenes menores de 20 años (183). Comienzo insidioso. La oftalmoplejía y la ptosis preceden a la proptosis y al desplazamiento del globo. La necrosis hemorrágica puede simular una osteomielitis. Síntomas de fatiga, anorexia, pérdida de peso y fiebre se asocian con metástasis sistémicas presentes entre el 10-30% de los pacientes al momento del diagnóstico (184). Se tratan con quimioterapia y radioterapia inicialmente. La cirugía se reserva para un segundo estadio. Combinando esas 3 técnicas se consigue un 53% de supervivencia a los 5 años y las metástasis disminuyen a un 5% (183,184).
TUMORES DEL NERVIO ÓPTICO Y DE DIFERENCIACIÓN NEURAL Gliomas del nervio óptico Suelen ser benignos y aparecen como media a los 9 años. Gliomas malignos son muy raros y prácticamente no hay en la infancia. A menudo son bilaterales (185) y afectan por igual a chicos y niñas. Según Dutton (29) la alteración primaria orbitaria es en un 24%, el quiasma en un 76% y el cerebro medio y tercer ventrículo en un 46%. 25-50% de ellos se asocian con neurofibromatosis (186-189).
Proptosis axial gradual y unilateral, no dolorosa, con pérdida progresiva de visión y defecto pupilar aferente son sus características clínicas. La atrofia óptica dobla en frecuencia al edema de papila. En la mayoría de los casos son autolimitados y crecen muy lentamente. Hay autores que los consideran hamartomas. Transformaciones quísticas de las lesiones pueden hacerlos crecer sin que se demuestre crecimiento celular (190).
Los gliomas ópticos se autolimitan en su crecimiento dentro del nervio sin invadir la duramadre, mientras que los asociados a neurofibromatosis proliferan en el espacio subaracnoideo siendo menos fusiformes (191).
El TC y la RNM delimitan la extensión y características de la invasión de canal óptico y quiasma, teniendo la RNM más definición. Es innecesario realizar una biopsia de los gliomas, aunque la anatomía patológica es definitiva. Hay falsos diagnósticos con meningioma debido a una hiperplasia meníngea reactiva alrededor de la tumoración, teniendo que profundizar en la biopsia para obtener la del glioma. Hay mucha controversia en el tratamiento de los gliomas de nervio óptico (189). Cada caso debe ser individualizado y las decisiones tomadas en función de las características de crecimiento del tumor, el tamaño y extensión en el nervio óptico, si está afectado el quiasma, etc.
Es importante la historia clínica, los tratamientos previos, la agudeza visual del ojo afecto y la del sano. Resaltar la evolución radiológica con controles periódicos de TC y RNM. Hay que evaluar alteraciones neurológicas, la edad de los pacientes, su proptosis, molestias, alteraciones estéticas y sistémicas (187). En el Hospital Infantil La Paz somos partidarios de observarlos con controles periódicos de neuroimagen mientras la agudeza visual sea buena. Hay muchos pacientes en la literatura que mantienen la visión durante años y nunca requieren cirugía. En los masivos para evitar la infiltración quiasmática se usa una orbitotomía transcraneal neuroquirúrgica. Se excinde el nervio óptico con liberación del canal y colocamos una prótesis integrada en la órbita suturándola a la musculatura extrínseca. El intentar conservar el ojo es una utopía si crecen rápidamente o son muy desfigurantes. Son casos límite la evidencia por RNM de estar a 3 mm del quiasma, el tener buena visión y la presencia de lesiones quísticas intratumorales que artefactan su real crecimiento. Sucede a veces que pese a la negatividad de las exploraciones complementarias, la invasión del quiasma se comprueba por anatomía patológica tras la cirugía y nos deja en mal lugar pese a los avances de la tecnología (187). La quimioterapia permite retrasar la necesidad de radioterapia con lo que las funciones endocrinas y su desarrollo cerebral son mejores, pero no es la panacea. La radioterapia externa es la última posibilidad cuando no se puede resecar, el tumor sigue creciendo a través de quiasma y cintillas ópticas y aparece sintomatología neurológica. Las complicaciones secundarias de la radiación en el crecimiento de estos niños y la aparición de segundos tumores son importantes (186). La mortalidad cuando llegan al quiasma es del 20% a los 10 años, en el cerebro del 55% y en la orbita del 5% (189). Las nuevas máquinas Gamma knyfe, LINAC (Acelerador lineal tridimensional) y Protones, parecen aportar nuevas terapéuticas pero actualmente hay muy poca experiencia en el ámbito mundial en oftalmología pediátrica.
Neurofibromas Tumoraciones de los nervios periféricos en párpados, orbita, SNC y vísceras. Durante la infancia sólo se manifiesta la neurofibromatosis tipo 1 que presenta un crecimiento plexiforme con afectación sistémica. Se conoce como enfermedad de Von Recklinghausen y es autosómica dominante con penetrancia incompleta. Es englobada en el grupo de las facomatosis y tiene múltiples características (manchas de café con leche, displasia de paredes orbitarias, glaucoma, gliomas del nervio óptico) (190).
En la cirugía los neurofibromas aparecen como gusanos tortuosos que infiltran los párpados y la órbita entre el tejido normal por lo que su extirpación total es muy complicada al implicar los fondos de saco y las conjuntivas, la glándula lagrimal y el elevador. Su resección implica afectar a estas estructuras por lo que hay que realizar suspensiones al frontal, injertos de mucosa para reconstruir los fórnix y cirugía plástica (191-193). Si además presentan una proptosis pulsátil por displasia esfenoidal, puede haber herniación cerebral. Las técnicas de reconstrucción con cirujanos maxilofaciales y craneales son bastante complejas y los resultados estéticos variables por la gran remodelación que precisan (193,194).
Un 15% de neurofibromatosis tipo 1 presentan gliomas del nervio ópticos control y evaluación es similar a las descritas en el apartado gliomas, pero en su pronóstico y dificultad quirúrgica son más complicados (195).
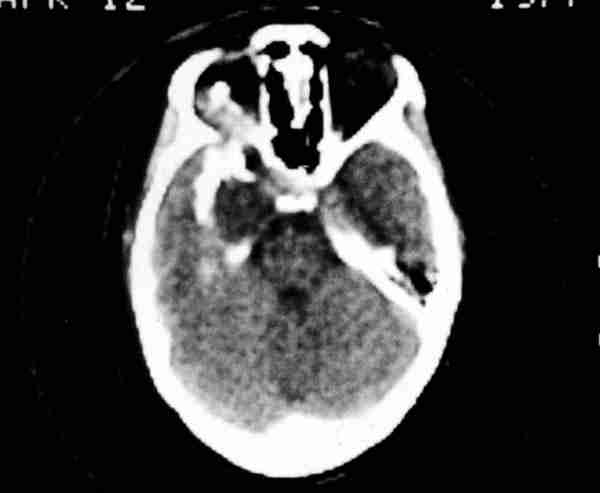
Meningiomas Son tumores que derivan de los plexos aracnoideos, siendo la mayoría intracraneales, en el surco olfatorio, ala mayor del esfenoides y supraselares, invadiendo la órbita secundariamente a través del hueso, canal óptico y hendidura esfenoidal (82,196,197). De 100 meningiomas sólo el 2,8% (2,2% de los tumores cerebrales en la edad evolutiva) corresponde a la infancia (196). Los primarios orbitarios también son muy infrecuentes y se forman en la aracnoides de la vaina del nervio óptico. Los bilaterales son raros a cualquier edad. En 9% de todos los meningiomas se asocian con neurofibromatosis (198,199). Defectos visuales, papiledema, proptosis, edema palpebral o quemosis son sus manifestaciones. El TC demuestra hiperóstosis y calcificaciones anormales intratumorales. La RNM con gadolinio delimita claramente su extensión pudiendo definirse el nervio óptico rodeado por la tumoración. Es la técnica de elección cuando el meningioma está confinado en la órbita y tiene buena agudeza visual (200). Otalmoplejía, defecto pupilar aferente y shunts opticociliares con distintas imágenes en la papila son típicos y dependen de la evolución (201).
El tratamiento como en los gliomas debe ser individualizado. La pérdida visual y la extensión intracraneal son factores importantes. Con radiación mejora la agudeza visual y se estabilizan (202,203). La cirugía es muy complicada y requiere mucha meticulosidad para extirpar el tumor sin dañar la visión. La quimioterapia no sirve para nada. Por suerte en niños no se dan meningiomas con proptosis invalidante y pérdida de agudeza visual (204). No precisan extirpaciones del tumor desde el globo ocular hasta el quiasma como sucede en los adultos. La supervivencia es excelente no existiendo en la literatura casos de muerte directa por el meningioma y sí por la cirugía. La mejoría de la agudeza visual tras radioterapia no compensa las complicaciones que puede producir en el desarrollo de los niños. Los hay que crecen hacia seno cavernoso y carótida siendo zonas de difícil acceso neuroquirúrgico y con posibilidades de producir iatrogenia (205,206).
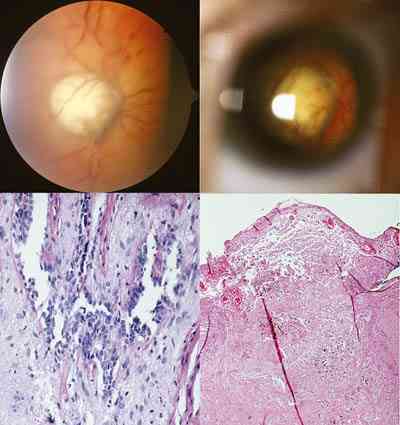
Meduloepiteliomas del nervio óptico Rara entidad tumoral definida como una neoformación constituida a partir de restos embrionarios del primitivo epitelio medular, que incluye estructuras derivadas de la vesícula óptica. Llama la atención revisada la literatura la mayor incidencia de distal a proximal en la localización de los dictiomas oculares. Así frente a los más de 70 casos descritos en la literatura mundial en cuerpo ciliar, disminuye bruscamente su incidencia en la localización retiniana y en papila sin afectación de la lamina cribosa, siendo rarísimos los de nervio óptico (207,209).
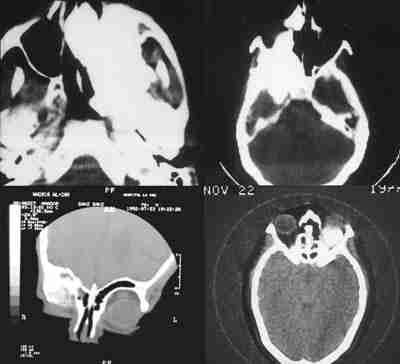
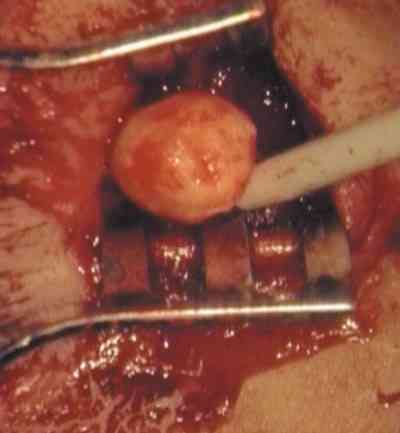
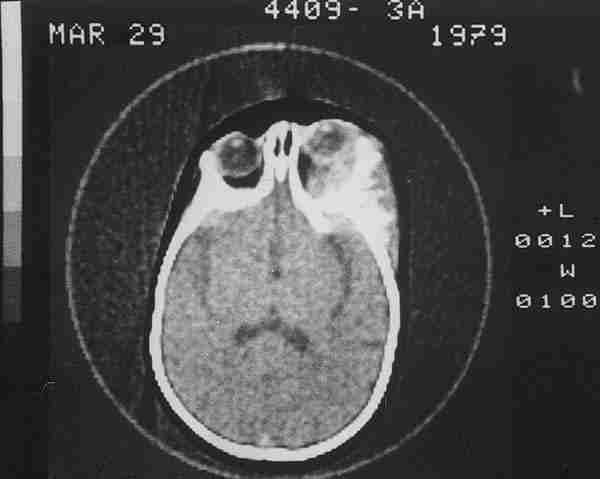
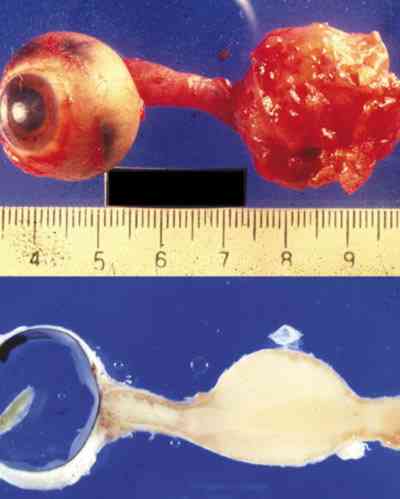
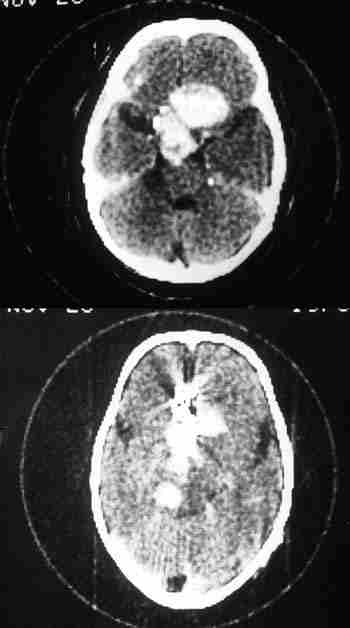
Sólo hemos encontrado 4 casos en la literatura mundial de estos últimos. Al Hospital Infantil La Paz nos remitieron en 1982 un niño de 12 años con una masa angiomatosa blanquecina-amarillenta que enmascaró en la papila durante mucho tiempo la presencia de un meduloepitelioma (dictioma) del nervio óptico. El curso clínico progresivo y la TC verificaron un diagnóstico de presunción hacia otro tipo de patología. La anatomía patológica tras biopsia realizada por neurocirugía, fue decisiva en el hallazgo tumoral que es el quinto existente en la literatura mundial y el primero en nuestro país de localización en el nervio óptico. Los casos encontrados en aquel tiempo eran muy pocos para predecir un pronóstico pese a su agresividad tumoral (210). La exenteración fue denegada por la familia y se le sometió a radioterapia. Tras 18 años de supervivencia, nuestro caso rompe todos los pronósticos existentes en la literatura.
Neurinomas (schwannomas) Hay series publicadas que los delimitan como el 6,8% de los tumores intracraneales, pero sólo el 1,1-5,7 de los intraorbitarios (82). Aparecen en niños por encima de los 8 años. Son proliferación de células de Schwan rodeadas por el perineuro de nervios orbitarios. Están bien encapsulados por lo que su extirpación es sencilla. En la literatura se manifiesta facilidad para la recurrencia incluso cuando la escisión fue completa. Pueden malignizarse esos restos (11,211-215). No se asocian con neurofibromatosis.
Tumor maligno periférico de las vainas de los nervios Neoplasia que en pacientes con neurofibromatosis plexiforme se desarrolla alrededor de los 10 años. También aparece espontáneamente o tras radioterapia, en la segunda década. Afecta al supraorbitario principalmente y puede invadir el cerebro por contigüidad dando metástasis a pulmones y ganglios linfáticos regionales. La aparición de dolor, hipoestesia y rápido crecimiento en pacientes con la enfermedad de Von Reklinghausen es muy significativa. Hay destrucción ósea y aumento de las hendiduras orbitarias. Pese a realizarles exenteraciones radicales, quimioterapia y radioterapia, el pronóstico vital es funesto. Antiguamente se clasificaba como neurinoma o neurilemoma maligno (215-219).
TUMORES NEUROEPITELIALES Tumor neuroectodérmico melanótico Tumor derivado de la cresta neural con marcada diferenciación hacia epitelio pigmentario y poca maduración del componente neuroblástico. Infiltra la órbita secundariamente desde el maxilar. Hay descrita afectación primaria. Es benigno y se reseca sin problemas (220,221). Con PAAF Galera-Ruiz ha presentado en 1999 un artículo de diagnóstico preoperatorio sobre este raro tumor de la edad neonatal (222).
Tumor neuroectodérmico periférico (pPNET) El concepto de tumor neuroectodérmico primitivo está en constante evolución. En el momento actual ya no están confinados en el sistema nervioso central SNC (PNET) sino que han aparecido fuera de su primitiva localización y se denominan pPNET. Son tumores que habían sido incluidos dentro de los Ewing con los que comparten alteraciones genéticas y musculares con diferenciaciones neuronales. En el capítulo de Patología los consideran extremos polares de un mismo grupo tumoral. La inmunohistoquímica ha permitido diferenciarlos en estos últimos años subdividiéndolos en varios subgrupos. Los tumores ectodérmicos periféricos (pPNET) son rarísimos en la órbita. Según Kiratli en su revisión de 1999 sólo 5 casos se conocen en la literatura en edad pediátrica (223). No hay consenso acerca el mejor tratamiento de estos tumores. Los de localización orbitaria pueden tener afectación ósea y extensión extraorbitaria regional. No metastatizan como los de otras localizaciones por lo que su pronóstico es mejor. Pueden tratarse como los Ewing y se consideran más agresivos que los rabdomiosarcomas (224,225). En la cirugía nuestro caso parecía un rabdomiosarcoma y se siguió las pautas de biopsia/ex tirpación que utilizamos habitualmente. La posterior clasificación por immunohistoquímica definió la patología. Se trató como un tumor de Ewing y en 2 años de evolución no ha habido incidencias.
TUMORES HEMATOPOYÉTICOS Es una compleja y confusa miscelánea de lesiones de la órbita, anejos oculares y globo con interrelación en entidades, con clínica, histología, inmunología y genética molecular muy variada. La incidencia a medida que avanzamos en sus conocimientos es cada vez más frecuente, pero todavía no hemos descifrado los factores que la producen.
Han pasado muchos años y la interfase entre la hiperplasia linfoide y los linfomas pasa por una gran variedad de patologías intermedias con gran cantidad de manifestaciones que se escapan a este capítulo.
Linfomas Knowles y Jakobiec señalan en 1983 que nunca habían visto un linfoma en la órbita como parte de una enfermedad sistémica en niños. El único linfoma que aparece en la órbita es el de Burkitt y en la infancia se pueden localizar en África. Es un tumor linfocítico altamente indiferenciado que afecta el doble a los varones africanos, con localización maxilar y en la glándula lacrimal. Uni o bilateralmente la malaria y el virus de Epstein Bar se relacionan con esta patología africana (22,226). Las variedades no africanas son escasísimas en órbita. Sólo hay 2 ó 3 casos descritos en la literatura. J. Shields (228) presenta el caso de un niño americano diagnosticado de linfoma primario. La inmunosupresión, los trasplantes de médula ósea y el SIDA en la infancia pueden aumentar la incidencia de metástasis en la órbita. Responden bien a la quimioterapia con remisión prolongada en la mayoría. Algunos remiten espontáneamente. La radioterapia en dosis de 2.500-3.000 rads se emplea en los orbitarios con excelente control. Metastatizan con frecuencia en la órbita (227,228).
Tumores de células plasmáticas Infrecuente en la infancia por ser predominantemente una neoplasia de variada virulencia desde benignas y reactivas a malignas. Las lesiones orbitarias pueden ser la primera manifestación de la enfermedad sistémica. Proptosis dolorosa y ptosis con infiltración subconjuntival, afectación de los senos paranasales con destrucción ósea, celulitis orbitaria, diplopía y edema palpebral. Todos los familiares deben ser examinados oftalmológicamente (229-231).
El tratamiento quirúrgico debe ser realizado con precaución. La radioterapia muestra buena respuesta aunque los hay radiorresistentes. Asociada al mieloma múltiple cuando progresa a través de los ganglios linfáticos tiene una supervivencia de menos de 2 años (232,233).
Leucemias Son características las afectaciones oculares y orbitarias. Se aprecian por infiltración en casi todas las estructuras del ojo. En la órbita dan infiltraciones difusas. La alteración hematológica produce hemorragias y hematomas que enmascaran los procesos (233-236).
En las linfoblásticas se afecta predominantemente el nervio óptico. Las mieloblásticas pueden debutar con manifestación orbitaria dando el denominado Cloroma o Sarcoma granulocítico. Hay más frecuentemente afectación orbitaria en las agudas que en las crónicas (237-239). Por regla general las manifestaciones sistémicas y afecciones cutáneas no hacen preciso la realización de biopsias orbitarias. Con PAAF se diagnostican y el tratamiento con quimioterapia sistémica reduce las alteraciones en el ojo y la órbita. Hay que tomar muestras del centro de la lesión porque en la periferia los cambios inflamatorios secundarios pueden dar falsos negativos (240-242). Puede haber afectación osea. En la actualidad hay mejores expectativas de supervivencia si el tratamiento con quimioterapia precede a la aparición de infiltración leucémica en la médula ósea y sangre periféricas. Precisan trasplante de médula osea para completar su evolución. La radioterapia es la última posibilidad (243). La genética está desarrollándose en este tipo de tumores intentando mejorar el diagnóstico y el tratamiento (244).
Histiocitosis y TUMORES ÓSEOS
Hay localización solitaria (granuloma eosinófilo) con proliferación de histiocitos en varios tejidos en la primera década de la vida, lesión lítica del techo de la órbita o del ala del esfenoides e inflamación perilesional del tejido limítrofe (245). Aunque la manifestación orbitaria sea única, el seguimiento debe de ser minucioso porque pueden aparecer manifestaciones multiorgánicas con diseminación sistémica (246).
Por debajo de los 2 años la supervivencia se refiere en la literatura como del 50%. Después aumenta hasta el 87% (245). La confirmación histológica con PAAFS, raspado de la lesión, corticoides intralesionales o bajas dosis de radiación define la evolución. Se dan falsos positivos de malignidad y la resección en bloque con los neurocirujanos del hueso para mejor preservar los márgenes quirúrgicos es obligada. Al analizar las piezas se confirma el fallo diagnóstico de las biopsias in situ. Algunas veces se han descrito remisiones espontáneas (247,248).
Xantogranuloma juvenil También llamado nevoxantoendotelioma y es en su rareza una afectación que pocas veces tiene localización orbitaria. Se da en niños por debajo de los 2 años y afecta a la piel y al iris dando hemorragias en cámara anterior, glaucoma, dolor y fotofobias. Son proliferación de histiocitos preferentemente siendo una histiocitosis no Langerhans' y las características células de Touton están descritas en todos los tratados. Tienen tendencia a regresión espontánea. Se presentan con proptosis unilateral y en la mayoría de los casos antes de los 6 meses de vida afectando a la órbita y músculos extraoculares (249-251).
Corticoides sistémicos y radioterapia en dosis bajas son las elecciones de tratamiento si se agravan y no se aprecia remisión (252,253).
Tumores de glÁndula lacrimal En la fosa lagrimal el dermoide es la patología más diagnosticada de todas las exploraciones complementarias neurorradiológicas por paradójico que resulte (17). Clínicamente las dacrioadenitis como variantes de los pseudotumores son masas inflamatorias de etiología desconocida que conviene diferenciar con las neoplasias. Las proliferaciones linfoides, linfomas de bajo grado y asociaciones con patologías inflamatorias están muy poco definidas en la literatura y en la infancia (254). Las técnicas de neuroimagen delimitan los procesos pero hay diagnósticos erróneos que pueden resultar fatales. El tratamiento con corticoides y las biopsias para evitar enmascarar los linfomas pueden dar sorpresas con la aparición de otras tumoraciones Tumores epiteliales. El 50% son adenomas pleomorfos también llamados tumores mixtos benignos. La otra mitad son carcinomas y de éstos en el adulto los adenoquistocarcinomas son mayoría (255). El tumor epitelial primario es el más infrecuente de todos en la infancia y se da a partir de los 10 años.
El mixto benigno también conocido como adenoma pleomórfico, en los adultos debe ser resecado con su pseudocápsula intacta por el peligro de recidivas que pueden dar degeneraciones malignas. Desde el año 1979 todavía nadie ha rebatido la experiencia personal de John E. Wright contraindicando las punciones y biopsias. Lo lógico es intentar resecarlo in toto con cápsula y márgenes de seguridad. Como a veces se rompe la cubierta se puede mandar una muestra intraoperatoria y maniobrar dependiendo del resultado. Por desgracia hay extensiones microscópicas del tumor fuera de la cápsula y recurren si no se quita suficiente margen del tejido orbitario cicunferencial. En los niños hay división de opiniones y se preconiza biopsiar primero en los últimos años. El problema en las recidivas es su riesgo de degeneración maligna. Con todo es muy importante planificar interdisciplinariamente la operación con los diversos servicios hospitalarios en un primer abordaje, porque si las extirpaciones radicales se hacen en segundos tiempos o tras las recidivas el pronóstico es muy malo (256). El adenoquistocarcinoma de glándula lagrimal o cilindroma también es raro pero hay varios artículos en la literatura sobre la infancia en los que se muestra la tendencia a desarrollar rápidamente y erosionar el hueso, con dolor por invasión perineural. Rootman refiere que la ausencia de erosión no excluye la malignidad por la gran remodelación ósea que se puede ver en la niñez con crecimientos rápidos (257). Tellado en 1997 presenta 11 casos con una media de edad de 14 años (2-18) y mayoría niñas con mejores perspectivas de supervivencia. Su pronóstico pese a la quimioterapia, radioterapia y cirugías mutilantes interdisciplinarias con extirpaciones en bloque del macizo facial no da resultado. Aunque se combinen todos los procedimientos, el índice de supervivencia es malo pese a tener menor agresividad histológica que en los adultos (258).
TUMORES ORBITARIOS SECUNDARIOS Son los que se extienden a la órbita desde las estructuras contiguas como globo ocular (retinoblastomas), párpados (carcinomas), senos paranasales (mucoceles, mucopioceles, angiofibromas, carcinomas, sarcomas, displasias y osteomas) o cerebro (meningiomas). Algunos como los meningiomas, displasias y alteraciones óseas se han tratado brevemente y otras se incluirán dentro de las especialidades respectivas en otros capítulos (259).
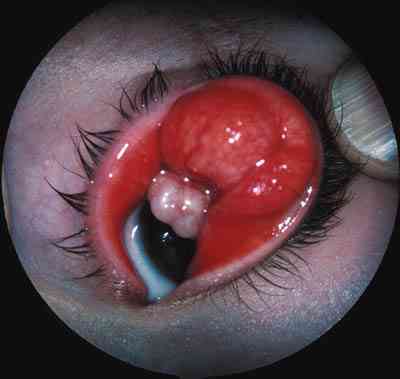
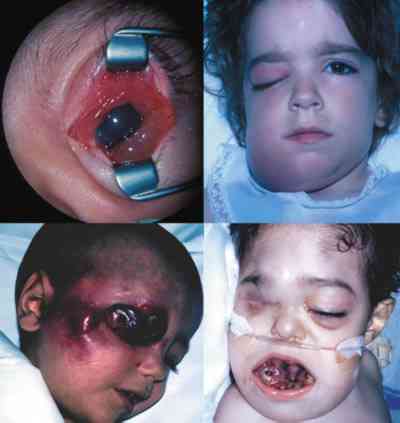
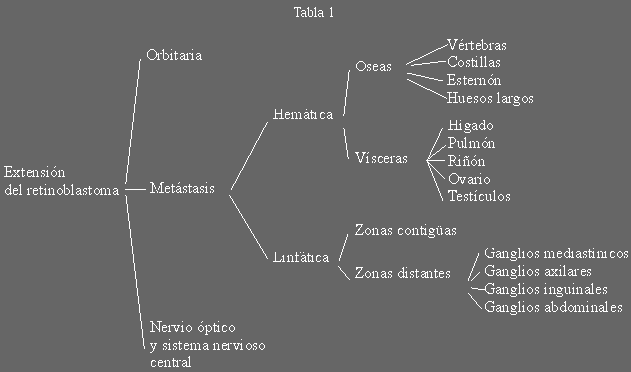
Extensión extraocular del retinoblastoma El retinoblastoma en el momento que atraviesa las envolturas oculares ha tenido a lo largo de los años un pronóstico vital muy malo, pese a la mejoría de las medidas terapéuticas empleadas. No hay sintomatología orbitaria si la extensión es microscópica. La importancia de un buen patólogo es fundamental para delimitar la infiltración del ojo (260). Las vías de extensión que debemos tener en mente al utilizar exploraciones complementarias con RNM cada 6 meses y TC cada 2 años en gran parte debido a la evolución y mantenimiento de los globos oculares gracias a la quimioterapia están descritas en la tabla 1 (261,262).
La genética es fundamental para el despistaje de portadores y pacientes con riesgo de desarrollar la enfermedad. Los estudios neurorradiológicos siempre deben incluir cortes craneales porque el pinealoblastoma debe estar presente en nuestras mentes pese a su rareza. Igualmente segundos tumores y tumores por radiación pueden aparecer durante toda su vida. No es aconsejable radiar antes del año de edad por el riesgo que representa. Debemos mantener con quimioterapia al niño durante meses para conseguirlo (263-265). Hay que mentalizar a la familia diciéndoles que los oftalmólogos controlamos la parte que nos corresponde, pero las revisiones oncológicas con estudios de neuroimagen, analíticos, etc. deben simultanearse por pediatras con práctica en tumores oculares y orbitarios. Cuando más seguros estamos y menos se piensa puede saltar la liebre (266-268). Igualmente participarles y que se involucren en el sinvivir de las revisiones. Que conozcan el riesgo que representa intentar conservar la poca agudeza visual que tienen cuando al ser bilaterales, hemos agotado todas las medidas terapéuticas y nos vemos en la obligación de enuclear el ojo con mejor agudeza visual (269).
Estéticamente una prótesis interna vascularizable (270) es preferible a un ojo amaurótico radiado cuando da problemas en su control. En Estados Unidos el porcentaje de supervivencia en series publicadas es del 90-95%. Afortunadamente aparecen resultados esperanzadores en otros países donde las condiciones médicas no son tan buenas. Kiratli presenta en 1998 (271) un estudio sobre 16 casos límite de retinoblastoma, tratados agresivamente con exenteración orbitaria, quimioterapia y/o radioterapia con magníficos resultados. Estas técnicas tienen absoluta vigencia hoy en día, pero si la extensión en la órbita es masiva las posibilidades de metástasis y mal pronóstico vital son muy elevadas. Se están desarrollando implantes intraorbitarios radiactivos en las recidivas y las nuevas técnicas de estereotaxia con LINAC, Gamma Nyfe y Protones usando el TC helicoidal pueden tener un gran futuro.
Melanomas Sólo 2 casos en la literatura con afectación orbitaria por melanomas de uvea son referidos en la literatura en edad neonatal (272). Broadway presenta un caso dramático de extensión extraocular y melanoma diseminado (273). Según Barr en su serie de 42 pacientes obtuvo un 25% de mortalidad. Los factores de mal pronóstico son semejantes a los de los adultos (274).
TUMORES METASTÁSICOS Neuroblastomas Es un tumor embrionario de la cresta neural que aparece en cualquier lugar del sistema nervioso simpático con un comportamiento variable. Puede crecer rápidamente, regresar o incluso madurar a ganglioneuroma. Requiere un completo examen oncológico y exploraciones complementarias por su diversificacion. La primera manifestación orbitaria a veces es una proptosis brusca y equimótica. La aparición de sangre en el párpado inferior da la impresión equivocada de traumatismo. Hay destrucción ósea. Los neuroblastomas congénitos de la cadena simpática cervical pueden producir un Horner homolateral con heterocromía (275-277). La biopsia orbitaria a veces es innecesaria por ser la clínica y la especificidad de los estudios sistémicos e inmunohistoquímicos definitivos. Con todo es obligatoria una confirmación histopatológica aunque sea de otras estructuras diferentes a las orbitarias (278,279). La órbita y los párpados son afectados bilateralmente a veces dando una imagen similar a los ojos de un oso panda. El pronóstico es muy malo pese a utilizar todas las posibilidades de quimioterapias, radioterapia y transplantes de médula ósea (280-282).
BIBLIOGRAFÍA
|