REVISIÓN
Dres. Villafruela Güemes IM.ª1, Tejada Palacios P2, García Silva M.ªT3, Arenas J4, Campos Y5
Servicio
de Oftalmología Pediátrica del Hospital 12 de Octubre. Madrid.
España.
(1) Licenciada en Medicina y Cirugía. MIR de Oftalmología.
(2) Doctora en Medicina y Cirugía. Médico Adjunto de
Oftalmología.
(3) Doctora en Medicina y Cirugía. Médico Adjunto de
Pediatría.
(4) Doctor en Medicina y Cirugía. Adjunto de Bioquímica.
(5) BSC. Investigadora.
Introducción
Las miopatías mitocondriales representan un grupo heterogéneo de enfermedades (1,2) que se asocian a defectos en el metabolismo energético mitocondrial.
En la biopsia muscular se pone de manifiesto las alteraciones existentes a nivel mitocondrial: anomalías morfológicas: fibras rojo rasgadas (ragged red fibers) que corresponden a acúmulos de mitocondrias con anomalías estructurales cuando se tiñen con tricrómico de Gomori, anomalías bioquímicas: déficits enzimáticos en la cadena respiratoria mitocondrial; y anomalías genéticas (3,16,18) con mutaciones del DNA mitocondrial humano.
La finalidad del metabolismo de la mitocondrial (10) es producir energía en forma de moléculas de ATP mediante reacciones de oxidación-reducción secuenciales localizadas en la matriz y en la membrana interna de la mitocondria.
La mitocondria, además, posee una composición genética peculiar, puesto que cada mitocondria posee su propio DNA mitocondrial (DNA mt) distinto del DNA nuclear con el que se sintetizan trece de las subunidades de los complejos enzimáticos de la cadena respiratoria.
Las miopatías mitocondriales siguen un patrón de herencia materno, no mendeliano, por ser el ovocito el que cede la gran mayoría de mitocondrias en el momento de la fecundación. Este hecho explica que estas enfermedades se transmitan desde la madre a todos los hijos independientemente del sexo de éstos.
El cuadro clínico que presentan los enfermos depende de numerosos factores (4): edad del paciente, distribución del defecto (especificidad del tejido y del grado de heteroplasmia mitocondrial en diferentes órganos) y el tiempo de evolución de la enfermedad.
Debido a la gran variedad de síntomas y signos (5) y su elevado grado de solapamiento, a veces, sólo la evolución de la enfermedad (8) puede dar las claves diagnósticas del fenotipo clínico (12).
La oftalmoplejia crónica externa progresiva (6) (CPEO) es un trastorno de la motilidad ocular extrínseca caracterizados fundamentalmente por una inmovilidad simétrica y lentamente progresiva de ambos ojos, siendo también típica la ptosis palpebral y una paresia del orbicular de los párpados. No se suele afectar la motilidad ocular intrínseca y no suele doler. En la infancia, normalmente comienza antes de los 5 años de edad, aunque algunos pacientes han sido diagnosticados a los 2 años o menores. Suele asociarse a deleciones del DNA mt o mutaciones.
Materiales y métodos
Nuestra casuística consta de 7 pacientes diagnosticados de miopatías mitocondriales sin signos neurológicos predominantes. Fueron remitidos desde la consulta de Enfermedades Mitocondriales para su exploración oftalmológica. A todos se les realizó según la colaboración y disponibilidad de medios un protocolo ofalmológico consistente en: agudeza visual, reflejos pupilares, existencia de tropias o forias, motilidad ocular extrínseca, refracción bajo cicloplejia, biomicroscopia, fondo de ojo tras midriasis farmacológica y pruebas electrofisiológicas.
Entre los 7 pacientes estudiados, un caso tenía ptosis palpebral, y otro caso tenía ptosis y oftalmoplejia crónica externa progresiva (11) (CPEO), 2 tenían una miopatía aislada y 3 casos tenían una miopatía asociada a otra enfermedad extraneurológica.
Resultados
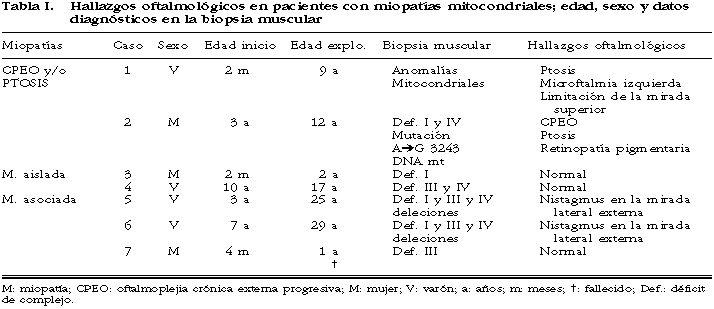
En este grupo de miopatías mitocondriales sin encefalopatías se han estudiado 7 pacientes de los cuales 4 son varones y 3 son mujeres (tabla I).
A continuación se describen los pacientes de nuestra serie con los hallazgos clínicos más característicos:
Caso 1: Es un niño que debutó con ptosis palpebral y microftalmia del ojo izquierdo en los primeros meses de vida.
En la biopsia muscular se encontraron anomalías estructurales de las mitocondrias.
En la exploración oftalmológica se apreció una ptosis bilateral que en el ojo izquierdo rebasa el área pupilar, con movimientos sacádicos de elevación palpebral. Presentaba una discreta limitación de la mirada superior y microftalmia bilateral más llamativa en el ojo izquierdo. La refracción era miópica. Con la biomicroscopia se apreció una foliculosis conjuntival en ambos ojos. En el fondo de ojo las papilas eran de aspecto normal y en la retina periférica temporal del ojo izquierdo se vieron algunos vasos envainados con aumento de la densidad interfase vitreorretina. Los potenciales evocados y el electrorretinograma realizados fueron normales.
Caso 2: Es una paciente que debutó a los 3 años de edad con una ptosis bilateral y progresiva. Además de presentar un déficit de los complejos enzimáticos I (NADH coenzima Q reductasa) y IV (citocromo C oxidasa) de la cadena respiratoria, tiene una mutación en el DNAmt (A > G 3243, en el tRNA leu) (20).
En la exploración oftalmológica destacó una ptosis bilateral que en el ojo derecho rebasaba claramente el reborde pupilar, y en el ojo izquierdo sólo lo rebasaba ligeramente, con una limitación importante de toda la motilidad ocular extrínseca. La motilidad ocular intrínseca era normal. Existía una exoforia al disociarla. En el fondo de ojo se apreciaron unas papilas discretamente pálidas con reborde peripapilar sobreelevado, de bordes netos y algo oblicuas, además presentaba tortuosidad vascular posterior en ambos ojos. Asimismo, tenía por fuera de arcadas un patrón pigmentario de aspecto granujiento y se veía mucho más la vascular coroidea. El electrorretinograma resultó poco valorable.
Los 2 pacientes con miopatía aislada sin encefalopatía ni retraso psicomotor eran una niña de 2 años de vida y un niño de 17 años.
Caso 3: Es una niña con un síndrome de hipotonía que ha mejorado con la edad.
En la biopsia muscular se demostró un déficit del complejo enzimático y de la cadena respiratoria.
La exploración oftalmológica era rigurosamente normal.
Caso 4: Es un niño que debutó a los 10 años de edad con debilidad muscular ligera, dolores y calambres musculares en los ejercicios prolongados.
Se encontró en la biopsia muscular un déficit de los complejos enzimáticos III (coenzima Q citocromo C reductasa) y IV de la cadena respiratoria.
La exploración oftalmológica resultó normal.
En el grupo de los pacientes con miopatía asociada a otros síntomas extraneurológicos, se incluyeron 3 pacientes, de los cuales 2 eran hermanos de 25 y 29 años de edad respectivamente y el tercer caso era una niña que murió al año de vida.
Caso 5 y 6: Son dos hermanos con un cuadro clínico muy similar, una miopatía sintomática desde la infancia, asociada a una anemia sideroblástica severa.
Además de un déficit plurienzimático (complejos I, III y IV de la cadena respiratoria) tienen deleciones en el DNA mt (19).
En la exploración oftalmológica lo único a destacar fue la presencia en ambos de un nistagmus de fijación en la mirada lateral.
Caso 7: Es una niña que murió al año de vida en la que se evidenció una miopatía mitocondrial junto a una miocardiopatía.
En la biopsia muscular se demostró un déficit del complejo enzimático III de la cadena respiratoria.
La exploración oftalmológica era compatible con la normalidad.
Discusión
La exploración oftalmológica en nuestra serie fue diferente según si los pacientes tenían una miopatía aislada o asociada a otra afectación extraneurológica o si presentaban o no oftalmoplejia externa progresiva (CPEO) y/o ptosis.
Si no tenían CPEO o ptosis la exploración oftalmológica era normal, salvo en dos casos que son hermanos y presentaron un nistagmus leve en la mirada lateral. Estos pacientes pueden evolucionar en un futuro hacia una CPEO. Es importante su seguimiento oftalmológico ya que tienen deleciones en el DNA mt, hallazgo frecuentemente asociado a ptosis o CPEO.
Una paciente con ptosis y CPEO presentaba una retinopatía pigmentaria con agudeza visual conservada pero sin ningún otro hallazgo que la englobara en un síndrome de Kearns-Sayre. El síndrome de Kearns-Sayre (7,14) se define clínicamente por la triada: CPEO, ptosis y retinopatía pigmentaria junto a uno de los criterios siguientes: alteraciones de la conducción cardíaca, hiperproteinorraquia o un síndrome cerebeloso.
La retinopatía pigmentaria descrita en las enfermedades mitocondriales se denomina atípica y puede cursar con uno de estos tres patrones:
1. La retinopatía más frecuente que encontramos en las enfermedades mitocondriales, como es el paciente de nuestra serie, es un fondo de ojo moteado hipopigmentado e hiperpigmentado como en sal y pimienta, asociado a una buena agudeza visual por localizarse frecuentemente por fuera de arcadas vasculares.
2. Retinopatía con espículas óseas, atrofia óptica, vasos sanguíneos atenuados denominada retinitis pigmentosa-like. La agudeza visual suele estar muy disminuida.
3. Retinopatía con una gran disminución de la agudeza visual, con atrofia severa del epitelio pigmentario retiniano, coriocapilares y del nervio óptico.
En 1989 Moraes et al. (13) estudiaron la presencia de deleciones del DNA mt en biopsias musculares de 123 pacientes, concluyendo que todos los pacientes con deleciones en el DNA mt presentaban CPEO y «fibras rojo rotas», este hecho fue confirmado por otros autores (15). La CPEO se asocia también con duplicaciones del DNA mt (16) y mutaciones del nucleótido en la posición 3243, típica del MELAS (9,17,18) (encefalopatía mitocondrial con acidosis láctica y accidentes cerebro-vasculares). En nuestra serie, una paciente presentaba CPEO y la mutación en la posición 3243 del DNAmt (tRNA leu).
Conclusiones
1. En los pacientes con clínica miopática aislada o asociada a síntomas extraneurológicos sin oftalmoplejia ni ptosis palpebral no se han encontrado manifestaciones oftalmológicas anormales.
2. Los pacientes con alteraciones pigmentaria retinianas se asocian a encefalopatías o a oftalmoplejia crónica externa progresiva (CPEO) por lo que requieren un estudio multisistémico más completo.
3. La presencia de retinitis pigmentaria en un paciente menor de 20 años con un cuadro de CPEO sugiere que puede haber en un futuro otra afectación sistémica o desarrollar un cuadro encefalopático, por lo que se debe realizar un seguimiento periódico.
Resumen
Las encefalomiopatías o enfermedades mitocondriales son entidades poco frecuentes que afectan a la totalidad del organismo y principalmente a los sistemas con una gran demanda energética como el cerebro, el músculo, la retina y el corazón. Están originadas por una alteración el metabolismo energético mitocondrial, demostrado mediante biopsia muscular.
Se describen las manifestaciones oftalmológicas de una serie de 8 pacientes diagnosticados de miopatías mitocondriales sin clínica neurológica predominante: un paciente presenta sólo ptosis palpebral, otro ptosis y oftalmoplejia crónica externa progresiva (CPEO), 2 pacientes miopatías aisladas y 3 pacientes miopatías asociadas a otra clínica extraneurológica.
Palabras clave
Miopatía mitocondrial, oftalmoplejia crónica externa progresiva (CPEO), retinopatía pigmentaria.
Abstract
The mitochondrial encephalomyopathies are rare diseases that may affect multiple systems and above all the central nervous system, skeletal muscle, retina and heart. They are produced by abnormalities in the mitochondrial energetic metabolism, as we can see on skeletal muscle biopsy.
The clinical features of 8 patients with mitochondrial myophaty without neurologic manifestations are described: a patient with ptosis alone, another patient with ptosis and chronic progressive external ophthalmoplegia (CPEO), 2 patients with only myophathy and 3 patients with myopathy and other extraneurologic disease.
Key words
Mitochondrial myopathies, chronic progressive external ophthalmoplegia (CPEO), pigmentary retinopathy.
Bibliografía
1. DiMauro S, Moraes CT: Mitochondrial encephalomyopathies. Arch Neurol 1993; 50: 1.197-1.207.
2. Johns DR: Mitochondrial DNA and disease. Seminars in medicine of the Beth Israel Hospital, Boston. N Engl J Med 1995; 33: 638-644.
3. Holt IJ, Harding AE, Cooper JM, Schapira AHV, Toscano A, Clark JB, MorganHughes JA: Mitochondrial myopathies: clinical and biochemical features of 30 patients with major deletions of muscle mitochondrial DNA. Ann Neurol 1989; 26: 699-708.
4. GarcRa Silva MT: Citopatías mitocondriales: aspectos clínicos. Rev Neurol 1994; 22: 182-185.
5. Petty RKH, Harding AE, Morgan-Huges JA: The clinical features of mitochondrial myopathy. Brain 1986; 109: 915-938.
6. Kosmorsky G, Johns DR: Neuro-ophthalmologic manifestations of mitochondrial DNA disorders: chronic progressive external ophthalmoplegia, Kearns-Sayre syndrome, and Leber's hereditary optic neuropathy. Neurol Clin 1991; 9: 147-161.
7. McKechinie NM, King M, Lee WR: Retinal pathology in the Kearns-Sayre syndrome. Br J Ophthalmol 1985; 69: 63-75.
8. Chang TS, Johns DR, Walker D, Maumenee IH, Green R: Ocular Clinicopathologic Study of the Mitochondrial Encephalomyopathy Overlap Syndromes. Arch Ophthajmolol 1993; 111: 254- 1.262.
9. Fang W, Huang C, Lee C, Cheng S, Pang C, Wei Y: Opthalmologic manifestations in MELAS syndrome. Arch Neurol 1993; 50: 977-980.
10. Pardo J, Prieto J: Enfermedades mitocondriales en Neurología. Medicine 1994; 6: 2.295-2.305.
11. Takeda S, Ohama E, Ikuta F: Involvement of extraocular muscle in mitochondrial encephalomyopathy. Acta Neuropathol 1990; 80: 118-122.
12. Mullie MA, Harding AE, Petty RKH, Ikeda H, Morgan-huges JA: The retinal manifestations of mitochondrial myopathy: a study of 22 cases. Arch Ophthalmol 1985; 103: 1.825-1.830.
13. Moraes CT, DiMauro S, Zeviani M, Lombes A, Shanske S, Miranda AF: Mitochondrial DNA deletions in progressive external ophthalmoplegia and Kearns-Sayre syndrome. N Engl J Med 1989; 320: 1.293-1.299.
14. Herzberg NH, Van Schooneveld MJ, Bleeker-Wagemakers EM, Zwart R, Cremers FPM: Kearns- Sayre syndrome with a phenocopy of choroideremia instead of pigmentary retinopathy. Neurology 1993; 43: 218-221.
15. Berners SM, Bacino C, Prezant TR, Pearson MA, Wood TS, Fournier P, Fischel-Ghodsian N: Identical mitochondrial DNA deletion in mother with progressive external ophthalmoplegia and son with Pearson marrow-pancreas syndrome. J Pediatr 1993; 123: 598-602.
16. Poulton J: Duplications of mitochondrial DNA: implications for pathogenesis. J Inherit Metab Dis 1992; 15: 487-498.
17. Moraes CT, Ciacci F, Silvestri G, et al: Atypical clinical presentations associated with the MELAS mutation at position 3,243 of human mitochondrial DNA. Neuromuscul Disord 1993; 3: 43-50.
18. Shing G, Lott MT, Wallace D: A mitochondrial DNA mutation as a cause of Leber's hereditary optic neuropathy. N Eng J Med 1989; 18: 1.300-1.305.
19. Casademont J, Barrientos A, Cardellach B, et al: Multiple deletions of mt DNA in two brothers with sideroblastic anemia and mitochondrial myopathy and in their asymptomatic mother. Hum Genet 1994; 11: 1.945-1.949.
20. Campos Y, Bautista J, Gutiérrez-Rivas E, et al: Variable clinical expression associated with the mutation 3243np of mitochondrial DNA. J Inher Metab Dis 1994; 17: 634-635.