 Fig. 1
Fig. 1COMUNICACIÓN SOLICITADA
Dres. Muñoz Negrete FJ1, Rebolleda Fernández G1, Gutiérrez Ortiz C2
Departamento de Oftalmología. Unidad de Glaucoma.
Hospital Ramón y Cajal. Madrid.
(1) Doctor en Medicina y Cirugía.
(2) Licenciada en Medicina y Cirugía.
Introducción
Diversos estudios han demostrado que la apoptosis puede ser el mecanismo fundamental de muerte de las células ganglionares en el glaucoma. La base genética y molecular de la apoptosis se está desvelando de forma cada vez más precisa, lo que podría permitir, en un futuro no muy lejano, el desarrollo de tratamientos neuroprotectores que complementarían la terapéutica antiglaucomatosa clásica.
El término apoptosis fue descrito por Kerr en 1972 (1). Deriva del griego apo (fuera de) y ptosis (cayendo). Se trata de una muerte celular programada, un suicidio celular codificado genéticamente, que se activa cuando la célula no es necesaria por más tiempo o cuando ha sido lesionada de forma severa (2,3).
Mecanismos de muerte celular
Existen tres patrones de muerte celular:
La apoptosis puede ocurrir en condiciones fisiológicas o en condiciones patológicas: 1. Apoptosis Fisiológica:
2. Apoptosis en condiciones patológicas: Puede activarse de forma inapropiada
en enfermedades neurodegenerativas como Parkinson y SIDA, en neoplasias así como en
trastornos isquémicos como el infarto de miocardio, accidentes cerebrovasculares, etc.
Desde el punto de vista morfológico, la apoptosis presenta cuatro fases (2):
1. Fase de Precondensación. La inducción de los genes requeridos para la apoptosis determina la transcripción y translación activa en proteínas y enzimas.
2. Fase de Condensación. Se produce una reducción del volumen de núcleo y citoplasma, con condensación de la cromatina en el margen nuclear. Hay una pérdida de las interacciones entre la célula apoptótica y las vecinas.
3. Fase de Fragmentación. Las proteínas de la matriz nuclear se solubilizan, lo que hace al DNA susceptible de degradación por endonucleasas dependientes de calcio y magnesio, que degradan la cromatina nuclear en los llamados cuerpos apoptóticos, que son múltiples y muestran un patrón en escalera en la electroforesis. Estos corpúsculos aumentan progresivamente en número hasta que el núcleo se hace picnótico.
4. Fase de Fagocitosis. Los cuerpos apoptóticos son fagocitados y degradados por las enzimas lisosomiales de las células vecinas sin respuesta inflamatoria alguna, por lo que no queda ni rastro de su presencia unas horas tras el comienzo del proceso.
Los dos criterios morfológicos fundamentales que definen la apoptosis son la degradación del DNA y la formación de nucleosomas en escalera. Sin embargo, no es fácil aislar células apoptóticas individuales, debido al pequeño número de células que sufren apoptosis y a la rapidez en que ésta ocurre (6-12 horas). Se ha desarrollado una técnica histoquímica denominada TUNEL ("Terminal Transferase dUTP-biotin Nick End-Labeling"), aunque tiene el inconveniente de no permitir en ocasiones diferenciar entre la formación de DNA en escalera y la degradación de DNA al azar que ocurre durante la necrosis, por lo que para una identificación exacta de la apoptosis se requiere la combinación de esta técnica y criterios morfológicos (7).
Relación entre apoptosis y patología oftalmológica
La apoptosis es un mecanismo de muerte celular en diversas enfermedades oculares (8). Se ha comprobado la presencia de apoptosis en las células ganglionares de la retina tras glaucoma experimental, sección del nervio óptico y en neuropatía óptica isquémica anterior. Asimismo se ha visto que pueda estar de alguna manera implicada en retinitis pigmentosa (9), desarrollo de catarata (10), retinoblastoma (11), isquemia retiniana y retinopatía diabética (8).
Control genético de la apoptosis
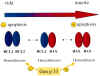
La mayor parte de nuestro conocimiento acerca del control genético de la apoptosis se basa en estudios de genes en el nematodo Caenorrhabditis elegans, cuyos genes de apoptosis han sido identificados y clonados, habiendo sido utilizada su secuencia de aminoácidos para la localización de los homólogos en humanos (12). Fruto de ello ha sido el aislamiento de genes de la familia bcl-2 (B-cell Iymphoma). Así se ha observado que el gen bcl-2 produce proteínas bcl-2, que bloquean la apoptosis en células ganglionares (13), mientras que el gen bax produce proteínas bax que estimulan la apoptosis. Se trata de proteínas diméricas, con secuencias de aminoácidos casi idénticas, que se unen entre sí formando heterodímeros. En condiciones normales, ambas proteínas se encuentran en un equilibrio dinámico, siendo la concentración de ambas similar o existiendo un mayor nivel de expresión de genes bcl-2, formándose homodímeros bcl-2 que protegen frente a la apoptosis (14). Si se produce un estímulo para la apoptosis, se sobreexpresan los genes bax o disminuye el nivel de proteínas de bcl-2, lo que determina la formación de homodímeros bax, que activan la apoptosis (fig. 1).
Fig. 1
Otro gen cuya expresión se ha observado incrementada en las células apoptóticas es el gen p53 supresor de tumor. Recientemente se ha observado que puede alterar el equilibrio bcl-2/bax, deprimiendo la transcripción de bcl-2 o activando la de bax (2). El gen p35 parece tener efecto inverso (15).
Estímulos desencadenantes de la apoptosis
Se ha postulado que los dos factores desencadenantes fundamentales de la apoptosis en las células ganglionares de la retina son la deprivación de factores neurotróficos y/o el incremento del nivel de excitotoxinas.
1. Deprivación de factores neurotróficos (neurotrofinas)
La mayoría de las neuronas dependen de factores de crecimiento para su supervivencia. Estos factores son pequeños péptidos (neurotrofinas, factores neurotróficos, citokinas o factores de crecimiento) que actúan uniéndose a receptores de la superficie de células diana, lo que desencadena una cascada de eventos moleculares, que suelen comenzar con la activación de la proteinkinasa y que afectan a múltiples funciones vitales del metabolismo de la célula (3). El más investigado es el BDNF (Brain derived neurotrophic factor), que es producido por las células cerebrales durante el desarrollo, siendo transportado por flujo axoplásmico retrógrado hacia la retina, por lo que las células ganglionares que fracasan en realizar sus conexiones con el cerebro mueren por ausencia de este factor, que es un componente esencial para su metabolismo. Este hecho explica la pérdida fisiológica de células ganglionares durante el desarrollo fetal.
La dependencia de las células ganglionares respecto al BDNF se mantiene a lo largo de toda su existencia, por lo que en la retina adulta cualquier mecanismo que impida el flujo retrógrado de BDNF (y/o otras neurotrofinas) compromete la viabilidad de las células ganglionares. Esto sucedería en la sección del nervio óptico, que ahora sabemos que estimula la apoptosis y que podría retrasarse mediante la inyección intravítrea de BDNF. Algo parecido aunque menos drástico ocurriría en el glaucoma, en el cual el aumento de la presión intraocular (PIO) interrumpiría el flujo axoplásmico anterógrado y retrógrado (16), lo que impediría la llegada de BDNF y estimularía la apoptosis (8). El mecanismo por el que podría actuar la deprivación de neurotrofinas podría ser la disminución en expresión de BCL-2.
2. Excitotoxinas y vía del óxido nítrico
La excitotoxina más estudiada es el glutamato, que en condiciones fisiológicas es un neurotransmisor excitador. Sin embargo, niveles excesivos resultan tóxicos y determinan la muerte de las células ganglionares retinianas (17). Olney (18) acuñó el término de excitotoxicidad para describir este efecto (Excitador+tóxico=Excitotoxina).
Una concentración local elevada de glutamato estimula receptores de la superficie celular, sobre todo NMDA (N-metil-D-aspartato), que abre los canales del calcio, determinando una sobrecarga intracelular de este ion, con la consiguiente activación de la enzima óxido nítrico sintetasa, que genera óxido nítrico, el cual posee propiedades de radical libre. Tanto glutamato como óxido nítrico son neurotransmisores en condiciones normales, pero cuando los receptores NMDA son hiperestimulados, el óxido nítrico se combina con intermediarios reactivos al oxígeno y genera peroxinitritos que producen la nitrosilación y fragmentación del DNA (fig. 2) (8,19).
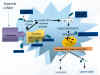 Fig. 2
Fig. 2
Se han encontrado niveles dos veces mayores de glutamato en vítreo de pacientes glaucomatosos respecto a pacientes con catarata aislada. Además existe una correlación entre la magnitud de incremento de glutamato y el tiempo de evolución del glaucoma (19). Estudios de cultivo celular han observado que son las células ganglionares grandes las que mueren de forma preferente cuando son expuestas a glutamato y NMDA, es decir las que primero se dañan en el glaucoma (20).
Mecanismo de elevación de glutamato a niveles tóxicos (Excitotoxina)
Dreyer (1998) postula tres teorías (19):
La toxicidad mediada por glutamato es una respuesta primaria a la isquemia celular. La deplección de energía determinaría un acúmulo de glutamato extracelular, que tras la hiperestimulación de receptores NMDA determinaría una sobrecarga de calcio intracelular. Por tanto, el glutamato podría jugar un papel importante en el mecanismo isquémico de daño del nervio óptico.
Parece que cuando la activación de los receptores de NMDA es leve se estimularía el mecanismo de la apoptosis, mientras que si la activación es intensa predominaría el mecanismo de la necrosis, por lo que podría ser que el glaucoma se deba a un estímulo leve a NMDA (21).
Estrategias neuroprotectoras y neurorregeneradoras
Basados en los mecanismos inductores de la apoptosis ya comentados podríamos establecer las siguientes líneas de tratamiento:
1. Terapia genética.
2. Introducción de factores neurotróficos.
3. Neuroprotección.
4. Neurorregeneración.
1. Terapia genética
Se podría bloquear la apoptosis intentando sobreexpresar genes bcl-2 en las células ganglionares. Así, en ratones transgénicos que sobreexpresan el gen represor de la apoptosis bcl-2 se ha observado un incremento de tamaño del nervio óptico y del tallo cerebral, aumento de espesor de las capas plexiforme interna y nuclear interna y aumento de células ganglionares en un 50% (22).
La dificultad en colocar DNA extraño dentro de las células del organismo y en conseguir que se mantenga un tiempo significativo (la mayoría de los métodos de transferencia genética resultan en expresión temporal de genes extraños) ha impedido la difusión de estas atractivas técnicas. Una opción que se está experimentando es utilizar vectores adenovirales benignos (23) o liposomas (24) para insertar DNA en las células retinianas.
La limitación principal de la terapia genética en el tratamiento del glaucoma es que se trata de una patología crónica, por lo que en el mejor de los casos la terapia genética proporcionaría sólo una solución temporal para una enfermedad prolongada.
2. Introducción de factores neurotróficos exógenos
Inyecciones de BDNF podrían retrasar la pérdida progresiva de células ganglionares. Pero antes de que pueda considerarse un tratamiento potencial habría que eliminar las serias complicaciones que presenta la administración sistémica de estos agentes. Un método muy prometedor, utilizado en la esclerosis lateral amiotrófica y cuyo uso podría plantearse en el glaucoma, consiste en la introducción de columnas de fibras poliméricas semipermeables con el centro hueco donde albergan células manipuladas genéticamente para segregar neurotrofinas (2).
3. Fármacos bloqueadores de la apoptosis (neuroprotectores)
Los objetivos de los fármacos neuroprotectores serían no sólo proteger los cuerpos celulares de axones dañados, sino también el rescate de axones no lesionados directamente, pero que pueden sufrir degeneración secundaria tras la liberación y extensión de sustancias nocivas desde la lesión primitiva.
Los fármacos neuroprotectores presentan dos inconvenientes muy importantes: En primer lugar, sólo producen protección parcial, ya que cada uno de ellos interfiere con un único evento desencadenante de la apoptosis. En segundo lugar, pueden interferir con funciones esenciales, que están mediadas por los mismos receptores que inducen daño secundario.
Fármacos neuroprotectores
Se han desarrollado unos trescientos antagonistas del glutamato (tabla I) (19). De entre ellos sólo comentaremos los más prometedores. Teniendo en cuenta el nivel de actuación dentro de la vía metabólica del glutamato, destacamos los siguientes (fig. 2).

1. INHIBIDORES DE LA LIBERACIÓN PRESINÁPTICA DE GLUTAMATO. El rilulazol ha sido aprobado en la esclerosis lateral amiotrófica.
2. ANTAGONISTAS de receptores NMDA. Las variantes de baja afinidad son las menos tóxicas, ya que permiten el mantenimiento de la actividad glutamatérgica basal. De ellos la memantina puede prevenir la muerte de células ganglionares mediada por activación de NMDA (25).
3. ANTAGONISTAS NMDA-like. Un analgésico no opiáceo, la flupirtina reduce el efecto de los radicales libres, protegiendo así a las células frente a la muerte celular por necrosis y/o apoptosis.
4. BLOQUEADORES DE LOS CANALES DEL CA. La Flunarizina favorece la supervivencia de células ganglionares tras la sección del nervio óptico en ratas adultas (26).
5. ANTIOXIDANTES. El 21-aminoesteroide mesilato de tirilazad bloquea la peroxidación de lípidos por radicales libres inhibiendo así la apoptosis (27).
6. ÁCIDO AURINTRICARBOXÍLICO. Previene la activación de la nucleasa que degrada el DNA (28).
7. BETAXOLOL. Además del efecto reductor de la PIO, mejora el flujo sanguíneo de la cabeza del nervio óptico y se le ha atribuido efecto neuroprotector por sus propiedades bloqueadoras de los canales del calcio tipo L.
8. BRIMONIDINA. Los a 2 agonistas son neuroprotectores en diversos modelos experimentales, como los de isquemia cerebral focal, aplastamiento calibrado de nervio óptico de rata, exposición excesiva a la luz, etc. El mecanismo neuroprotector podría estar relacionado con la producción de factores de crecimiento como el factor de crecimiento fibroblástico básico. Para que la brimonidina ejerza este efecto neuroprotector sería preciso que alcanzara niveles terapéuticos en el segmento posterior tras su aplicación tópica; así se ha observado que alcanza en vítreo una concentración 50-80 veces superior a la necesaria para activar los receptores a 2. Por otro lado, al igual que el timolol se une reversiblemente a la melanina ocular pudiendo actuar como depot de liberación sostenida en vítreo y retina neural (29).
Estos hallazgos sugieren un efecto beneficioso potencial en el tratamiento del glaucoma más allá de la simple reducción de la PIO.
4. Neurorregeneración
Su descripción detallada escapa de los límites de la revisión presente. Consistiría en inducir la regeneración de las neuronas cuyos axones han sido dañados, antes de que sus cuerpos celulares se atrofien. En contraste con las neuronas periféricas, la mayoría de las neuronas centrales maduras prácticamente no presentan regeneración axonal. Como dato curioso reseñaremos que las células ganglionares retinianas pueden regenerarse dentro de injertos del sistema nervioso periférico pero no dentro del SNC. En la tabla II se reseñan diversas técnicas de neurorregeneración.

En los injertos de nervios periféricos, un extremo del injerto es adherido al muñón de nervio óptico seccionado y el otro puede ser dejado sin conectar o guiado hacia diferentes localizaciones como tubérculos cuadrigéminos superiores o centros diana inadecuados para las fibras nerviosas ganglionares (cerebelo). Estos injertos sintetizan NGF (nerve growth factor), BDNF, CNTF (ciliary neurotrophic faclor) y FGF (fibroblast growth factor), pero esta capacidad la pierden con el tiempo por lo que sólo ejercen efectos temporales sobre la supervivencia de las células ganglionares y únicamente aquellas fibras que consiguen realizar conexiones sinápticas sobreviven.
Respecto a la administración de factores tróficos, el más potente es el BDNF. Tras su inyección intravítrea se retrasa la apoptosis de células ganglionares, aunque el efecto va disminuyendo con el tiempo conforme los niveles de BDNF se van agotando (8). Se está investigando si una suplencia continua de BDNF podría permitir el rescate de células ganglionares dañadas durante períodos más largos y en un número mayor. También se está investigando la utilidad de CNTF, FGF y NGF.
Conclusiones
La prevención de la muerte de las células ganglionares no es probable que reemplace los tratamientos habituales del glaucoma, ya que la apoptosis es sólo la evolución final de muchas enfermedades y no la causa. El bloqueo de la apoptosis sólo sería válido utilizado en combinación con tratamientos dirigidos hacia la causa principal. Sin embargo, podría conseguir que las células ganglionares se recuperaran de un período de stress inicial, permitiéndoles una recuperación completa si la causa primitiva (PIO elevada) es reducida y controlada. Por tanto, en el futuro la combinación de terapias neuroprotectoras y reductoras de la PIO podría convertirse en el tratamiento de elección del glaucoma.
Bibliografía