 Fig. 1
Fig. 1Dres. Gonzalvo Ibáñez FJ1, Playán Ariso A2, Izaguirre Roncal L1, Alcaine Villarroya MJ2, Fernández Tirado FJ3, Honrubia López FM3, Montoya Villarroya J4
(1) Licenciado en Medicina y Cirugía. Servicio de
Oftalmología. Hospital Miguel Servet. Zaragoza. España.
(2) Licenciada en Ciencias Químicas. Departamento de Bioquímica y Biología Molecular y
Celular. Universidad de Zaragoza. España.
(3) Doctor en Medicina y Cirugía. Servicio de Oftalmología. Hospital Miguel Servet.
Zaragoza. España.
(4) Doctor en Farmacia. Departamento de Bioquímica y Biología Molecular y Celular.
Universidad de Zaragoza. España.
Proyecto subvencionado PB-94-0567 de la Dirección General de Investigación Científica y Técnica y P24/97 de la Diputación General de Aragón.
Introducción
Las citopatías o encefalomiopatías mitocondriales son un grupo heterogéneo de enfermedades caracterizadas por una alteración primaria del metabolismo mitocondrial. El síndrome de Kearns-Sayre (SKS) ocupa una situación central dentro del amplio espectro de las encefalomiopatías mitocondriales. Se trata de un cuadro con afectación multiorgánica definido por: inicio antes de los 20 años de edad, oftalmoplejía externa crónica progresiva (OECP), retinopatía pigmentaria y al menos, uno de los tres siguientes criterios: alteraciones en la conducción cardíaca, disfunción cerebelosa e hiperproteinorraquia >100 mg/dl. Otros signos neurológicos y endocrinológicos pueden asociarse al SKS.
La primera descripción de deleciones en el ADN mitocondrial (ADNmt) en pacientes con miopatías mitocondriales definidas por criterios clínicos e histológicos fue realizada por Holt et al en 1988 (1). Los estudios del ADNmt en pacientes con SKS han demostrado la presencia de diversas alteraciones, entre las que se incluyen deleciones parciales, con diferentes porcentajes del total de ADNmt delecionado y duplicaciones (2,3).
Presentamos los datos clínicos y resultados del estudio de deleciones parciales en el ADNmt de una paciente diagnosticada de SKS (4).
Caso clínico
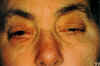
Mujer de 63 años de edad, sin antecedentes familiares significativos. En la exploración oftalmológica se encontró una agudeza visual de 0,3 en AO, ptosis bilateral de inicio a los 17 años en OD y 19 en OI, de carácter progresivo (fig. 1), oftalmoplejía completa progresiva de inicio a los 30 años, que es de carácter total desde los 46 años; en ningún momento de la evolución presentó diplopía y las pupilas persistieron isocóricas y normorreactivas. El fondo de ojo reveló una palidez papilar y una retinopatía pigmentaria en AO, con movilización pigmentaria a partir del ecuador con pseudoespículas óseas en la periferia retiniana (figs. 2 y 3). Se encontraron reducciones de la amplitud de la onda b del electrorretinograma y del componente P100 del potencial evocado visual.
Fig. 1
 Fig. 2
Fig. 2
 Fig. 3
Fig. 3
A nivel sistémico presentó los siguientes hallazgos: marcapasos endocavitario definitivo por disfunción sinusal y bloqueo auriculoventricular infrahissiano. Intolerancia al ejercicio y debilidad muscular progresiva, más acentuada en cintura escapular, con patrón miopático en el electromiograma de músculos deltoides y orbicular de párpados. Hiperproteinorraquia (170 mg/dl) con normalidad en el resto de parámetros bioquímicos, estudio citológico, microbiológico e inmunológico del LCR. CI=79 en el test WAIS (Wechsler Adults Intelligence Scale). Hipoacusia bilateral perceptiva del 70%, exclusiva para 4.000 c/s. Aumentos del lactato (37,92 mg/dl) y piruvato (1,12 mg/dl) plasmáticos. Aclaramiento plasmático de Fe59 radiactivo acelerado y depósito anómalo de Fe59 en áreas basales cerebrales. TAC cerebral normal.
El estudio endocrinológico (gammagrafía y hormonas tiroideas, PTH, Ca, P, fosfatasa alcalina, hormonas hipofisarias, sexuales y sus metabolitos en orina, cortisol, catecolaminas y test de glucagón), el estudio microbiológico sanguíneo y los niveles séricos de transaminasas y alanina fueron normales. El estudio con microscopia óptica a nivel del músculo esquelético detectó alteraciones compatibles con miopatía mitocondrial, siendo las muestras de piel y nervio periférico normales.
Estudio genético: Cariotipo 46,XX. El análisis de deleciones del ADNmt se realizó mediante la técnica de Southern blot a partir de sangre venosa y músculo esquelético. El ADN total fue sometido a digestión por la enzima de restricción PvuII que linealiza el ADNmt, reconociendo como punto de corte la posición 2.652 del ADNmt. La separación de fragmentos se obtuvo mediante electroforesis en geles de agarosa al 0,7%. Tras una transferencia por vacío a membranas de nylon, se realizó la hibridación con una sonda de ADNmt obtenida por amplificación de un fragmento de ADNmt de la región 16S ARNr-ND1 y marcada con digoxigenina. Los fragmentos de ADNmt se detectaron por fluorescencia y se impresionaron en placa de rayos X con el fin de estudiar el patrón generado por la digestión del enzima. La cuantificación de las diferentes poblaciones de ADNmt se realizó mediante densitometría. Las localizaciones de los límites de la deleción se determinaron mediante amplificación del ADNmt por la reacción en cadena de la polimerasa, clonación de los fragmentos amplificados y secuenciación de los mismos (5). En la muestra de músculo esquelético se detectó la presencia de una deleción parcial con un tamaño de 4.977pb y con un porcentaje del 72% del ADNmt delecionado (fig. 4). La deleción se localizó entre los nucleótidos 8.483 a 13.459, con afectación de las regiones ATPasa 8 y 6, COII, ND3, ND4L y ND5 (fig. 5). En las muestras de sangre venosa de la paciente y de dos hijos y una hija, todos ellos asintomáticos, no se encontraron deleciones (fig. 4).
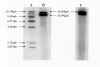 Fig. 4
Fig. 4
 Fig. 5
Fig. 5
Discusión
La afectación de la musculatura extrínseca ocular (OECP, o incluso sólo ptosis) y la intolerancia al ejercicio son los síntomas más frecuentes en los pacientes con enfermedad mitocondrial, siendo éstos los síntomas de presentación en el caso que nos ocupa (6). La OECP es el dato clínico más firmemente asociado a la presencia de deleciones en el ADNmt (7). La oftalmoplejía en nuestro caso se inició a los 30 años y, en general no suele presentarse antes de los 5 años de edad, no suele causar diplopía, respeta siempre las pupilas y puede progresar a una oftalmoplejía completa como ocurrió en nuestra paciente.
La retinopatía pigmentaria es un dato frecuentemente asociado a las miopatías mitocondriales, habiéndose definido tres patrones diferentes de presentación (8). Nuestra paciente se ajustó al tipo de retinopatía, caracterizada por cambios similares a los de la retinitis pigmentaria con formación de pseudoespículas óseas, atrofia óptica y atenuación vascular. En general, la retinopatía pigmentaria en el SKS no sigue un patrón típico de retinitis pigmentaria con afectación inicialmente periférica y centrípeta, sino que presenta afectación predominante del fondo central y aproximadamente sólo el 50% de los casos desarrollan síntomas visuales en forma de déficit visual moderado.
El modelo de herencia en el SKS no ha sido todavía establecido. La presentación esporádica es la más frecuente y la aparición de una mutación espontánea es la hipótesis más aceptada, pudiendo ésta ocurrir en el oocito o en el cigoto. Un reciente estudio ha confirmado la existencia de oocitos que contienen niveles detectables (superiores al 0,1%) de una deleción que afecta a 4.977pb, similar a la detectada en nuestra paciente (9).
Las deleciones en el ADNmt descritas en diferentes estudios van desde 1,3 a 7,6Kb (7). La deleción detectada en nuestra paciente y que afecta a 4.977pb entre las posiciones 8.483 y 13.459, es la más frecuentemente descrita en el SKS en diferentes series y ha sido también descrita en otras miopatías mitocondriales (7,10). Este hecho ha sugerido la existencia de puntos en el ADNmt con mayor predisposición a la deleción (hot-spots).
La proporción de moléculas de ADNmt delecionado ascendió al 72% en nuestra paciente, situándose próxima al límite superior del rango detectado en diferentes estudios (entre un 25 a 93% del total de ADNmt delecionado) (7,11). Aunque no se ha podido demostrar correlación entre la severidad clínica o anomalías bioquímicas con la localización y tamaño de las deleciones o con el número de genomas delecionados, se ha propuesto la existencia de un umbral mínimo en el número de moléculas de ADNmt delecionado para la expresión fenotípica de la enfermedad (efecto umbral), situado en el 18% de ADNmt delecionado (12).
Algunos estudios no han encontrado deleciones en el ADNmt leucocitario de pacientes con deleciones del ADNmt muscular (1,13). En nuestra paciente las deleciones fueron detectadas en músculo pero no en sangre. Sin embargo, ha sido descrita la presencia de deleciones del ADNmt en diferentes tejidos (músculo esquelético, linfocitos, hígado y cerebro) de un mismo paciente. Dichas deleciones eran idénticas en extensión y localización en los diferentes tejidos de un mismo paciente, pero la proporción de ADNmt delecionado fue siempre superior en músculo esquelético (7).
Conclusiones
1. El análisis del ADNmt puede ser útil en el diagnóstico del SKS, especialmente en aquellos casos que no presenten la totalidad de criterios diagnósticos.
2. La deleción del ADNmt que afecta a 4.977pb es la más frecuentemente detectada en el SKS.
3. El estudio de ADNmt debe realizarse en el tejido muscular afectado, ya que en sangre venosa pueden aparecer resultados negativos en pacientes con deleciones del ADNmt.
Resumen
El síndrome de Kearns-Sayre es una enfermedad poco frecuente perteneciente al grupo de las encefalomiopatías mitocondriales. Estas enfermedades presentan carácter sistémico con afectación predominante de los tejidos con alta demanda de energía: músculo esquelético, corazón, sistema nervioso central y retina.
Presentamos el caso de una mujer diagnosticada clínica y anatomopatológicamente de síndrome de Kearns-Sayre. El análisis de la posible presencia de deleciones en el ADN mitocondrial se realizó en muestras de músculo orbicular y sangre venosa mediante la técnica de hibridación Southern y secuenciación. En la muestra de músculo se detectó la presencia de una deleción parcial con un tamaño de 4.977 pares de bases y con un porcentaje del 72% del ADN mitocondrial delecionado. La deleción se situó entre los nucleótidos 8.483 y 13.459. En las muestras de sangre venosa de la paciente y de tres de sus hijos no se encontraron deleciones.
El estudio de deleciones del ADN mitocondrial es una prueba útil en el diagnóstico del síndrome de Kearns-Sayre. El estudio del ADN mitocondrial se debe realizar en el tejido muscular afectado, ya que pueden aparecer resultados negativos en sangre venosa en pacientes portadores de deleciones del ADN mitocondrial.
Palabras clave
Síndrome de Kearns-Sayre, ADN mitocondrial, deleción.
Summary
Kearns-Sayre syndrome is an unusual disease belonging to mitochondrial encephalomyopathies. Such diseases are multisystem disorders with a predominant dysfunction of organs with high energetic requirement: skeletal muscle, heart, central nervous system and retina.
We present a case of a woman with a clinical and anatomopathologic diagnosis of Kearns-Sayre syndrome. Orbicular muscle and venous blood samples were taken for the mitochondrial DNA analysis. Southern blot technique and sequencing studies showed the presence of a 4,977 base pair deletion in the muscle sample in a proportion of 72%. The deletion extended from nucleotids 8,483 to 13,459. No deletions were found in the venous blood samples of the patient and her son and daughter.
The study of mitochondrial DNA deletions is a helpful test in the diagnosis of Kearns-Sayre syndrome. It must be made in the affected muscle, since we may obtain negative results of venous blood samples in pathiens with mitochondrial DNA deletions.
Key words
Kearns-Sayre syndrome, mitochondrial DNA, deletion.
Bibliografía