REVISIÓN
Retinopatías externas focales agudas
Dr. Clement Fernández F
Servicio de Oftalmología. Hospital de la Princesa.
Madrid. España.
Introducción
En estos últimos años se han descrito una serie de enfermedades
coriorretinianas multifocales idiopáticas, de posible etiología inflamatoria y de
comienzo en general agudo que afectan a adultos jóvenes y con un claro predominio por el
sexo femenino. Estos pacientes no padecen enfermedades sistémicas, ni alteraciones
inmunológicas conocidas y según el aspecto que se ha destacado se han incluido en los
síndromes de puntos blancos destacando las manchas blanco-amarillentas visibles en el
polo posterior y media periferia, o síndromes de aumento idiopático de mancha ciega,
haciendo referencia al escotoma centrado en la papila que acompaña a varios de ellos. En
1993 Gass, acuñó el término de AZOOR (Acute zonal outer occult retinopathy),
agrupándolos desde un punto fisiopatológico por la afectación aguda y focal de la
retina externa .
Clasificación
Se incluyen dentro de este grupo los siguientes procesos:
- AIBSE Acute idiopathic blind spot enlargement (Fletcher, 1988).
- AMN Acute macular neuroretinopathy (Bos y Deutman, 1975).
- MEWDS Multiple evanescent white-dot syndrome (Jampol, 1984).
- PIC Punctate inner choroidopathy (Whatzke y Folk, 1984).
- CM -panuveitis - multifocal choroiditis (Nozik y Dorsch, 1973).
- DSFS - Difuse subretinal fibrosis syndrome.
- AZOOR- acute zonal occult outer retinopathy (Gass, 1993).
- ARPE - acute retinal pigment epithelitis (Deutman).
Algunos autores incluyen al AMPPE y la coroiditis serpiginosa en este gran grupo, así
como la maculopatía idiopática aguda unilateral (MIAU) descrita por Yannuzzi y cols en
1991 (1).
Como expresa en una editorial L. Jampol (2): "Esta sopa de letras es la
consecuencia de nuestro desconocimiento sobre la etiología y patogenia de estas
entidades. Mientras que no tengamos más información de su causa, la relación entre
estas entidades permanece incierta".
Aunque han sido descrito pacientes con cuadros mixtos (Mewds y Cm, Mewds y AMN, Aibse
con Mewds, Amn. Cm) (3), y tal vez como defienden algunos autores encabezados por Gass,
Ben Ezra y Forrester representen un espectro de repuestas de un mismo proceso etiológico,
las características clínicas de los mismos nos permiten establecer tres grupos
diferenciados (tabla I).

Clínica
I) CM -PIC-DSFS-ARPE
Estos cuadros clínicos afectan a adultos jóvenes miopes, con un claro predominio por
el sexo femenino que va desde 9/1 en el PIC a 3/1 en la CM y en general sanos, aunque en
la CM se han descrito procesos febriles previos y se ha asociado a diversas patologías
(herpes simple y zóster, mononucleosis, enf. de Lyme, sarcoidosis, síndrome de Reiter).
El cuadro es generalmente bilateral aunque puede ser asimétrico, y se caracteriza
clínicamente por la presencia de miodesopsias, fotopsias, escotomas o disminución de la
agudeza visual y en la oftalmoscopia se aprecia la presencia de focos amarillentos
subretinianos situados en polo posterior y periferia, de tamaño variable entre 50 y 1.000
micras y en número de 3 hasta un centenar, que evolucionan bien desapareciendo o
cicatrizando dejando focos en sacabocados con mayor o menor reacción pigmentaria
similares a las descritas en el POHS. Se puede asociar un discreto edema del disco óptico
(fig. 1).
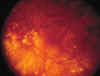 Fig. 1
Fig. 1
En la evolución, pueden presentarse membranas neovasculares coroideas maculares MNVC
en un 20 al 40% (fig. 2).
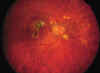 Fig. 2
Fig. 2
En la AFG, en la fase aguda dichos focos aparecen hipofluorescentes, con una
impregnación moderada en fases tardías, comportándose como efectos ventana en fase
cicatricial.
En la ICG, se caracterizan por la aparición de focos hipofluorescentes, en tiempos
medios y tardíos, con tendencia en la CM a agruparse entorno a la papila, asociándose a
veces a un aumento de la mancha ciega (4), sin alteraciones oftalmoscópicas papilares (5)
(fig. 3).
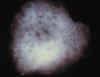 Fig. 3
Fig. 3
El ERG, suele ser normal en la CM, pudiendo ser normal o subnormal en el SDFS.
Características diferenciales
Existen sin embargo diferencias entre los cuatro que nos permiten realizar el
diagnóstico diferencial entre ellos:
La CM es un cuadro crónico por lo que podemos ver focos en estadios cicatriciales y
otros activos y presenta un tyndal vítreo positivo (en un 50% existe además un tyndal
celular anterior), mientras que en el PIC, el cuadro es agudo con muy raras recurrencias
tras los 2 ó 3 primeros meses, y el tyndal es en todos los casos descritos negativo (fig.
4).
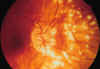 Fig. 4
Fig. 4
Cuando los focos están en fase cicatricial, la CM se asemeja a la pseudo y presunta
histoplasmosis ocular (PPHOS-POHS), no solo por las características de dichos focos sino
por la existencia de cambios pigmentarios peripapilares y estrías lineales periféricas
(6,7). Para diferenciar ambos cuadros es de utilidad la ICG (8) ya que a diferencia de los
focos hipofluorescentes visibles en la CM, en el POHS se comprueba además la existencia
de focos hiperfluorescentes. Asimismo en la CM el HLA-DR2 (9,10) es negativo a diferencia
del POHS, donde es frecuentemente positivo. Por fin en la CM así como en el PPHOS, no
existen anticuerpos frente al histoplasma capsulatum (fig. 5).
 Fig. 5
Fig. 5
En 1997, Charloteww y cols (11) propusieron dividir la CM en dos formas, la posterior o
típica, cuyas características hemos descrito y una forma periférica que se caracteriza
por afectar a mujeres de mayor edad (edad media 58 a, frente a edades entre 20 y 50 a en
la forma típica), y en la cual las lesiones se localizan sólo en la periferia, con
frecuente edema macular quístico (72%) (10 al 20% en la forma típica) y que rara vez se
complican con MNVC macular. Estas formas se complican frecuentemente de cataratas (62%) y
glaucoma (25%), y se asocian a sarcoidosis.
El DSFS, presenta algunas características propias, con un comienzo agudo unilateral
aunque en el 76% se bilateraliza. Los focos blanquecinos posteriores tienden a coalescer
formando una placa amarillenta con líquido subretiniano turbio, evolucionando a la
formación de una cicatriz fibroglial subretiniana macular; asimismo dicha reacción
fibroglial puede producirse en algunos de los focos periféricos (12). En el 77% presenta
un tyndal vítreo positivo y en el 66% en cámara anterior, pudiendo en algunos casos
presentar sinequias, posteriores, atrofia de iris y precipitados queráticos (13).
EL ARPE (14), afecta por igual a ambos sexos y se caracteriza por la aparición de
manera uni o bilateral, de focos finos grisáceos con un halo amarillento en torno,
localizados en polo posterior. En fase aguda el EOG es anormal, normalizándose
posteriormente, mientras que el ERG y PEV son normales. La recuperación clínica es total
en 7 a 10 semanas, persistiendo las pequeñas lesiones pigmentadas.
Tratamiento
El empleo de corticoides perioculares o sistémicos suelen ser eficaces en la CM en los
primeros estadios, pero tiende a hacerse refractaria a los mismos, por lo cual debe
valorarse el empleo de inmunosupresores asociados. Nolle 1998 (15) encuentra así mismo un
mal pronóstico tanto en la cirugía de cataratas con frecuente rubeosis y glaucoma como
en la vitrectomía. En la DSFS, el pronóstico visual es malo a pesar del tratamiento,
siendo preciso el empleo de corticoides a altas dosis (prednisona 80 mg/d) en fase precoz
ya que cuando la fibrosis se ha iniciado la respuesta terapéutica es pobre.
En caso de aparecer una MNVC, si ésta es extrafoveal se puede realizar tratamiento con
corticoides y láser. En las formas subfoveales (16) la cirugía de dichas membranas no ha
obtenido resultados visuales mejor que con la corticoterapia, siendo la tasa de
recurrencias alta.
II) MEWDS
Esta entidad fue descrita en 1984 por Jampol, afecta a jóvenes
adultos sanos (aunque en los casos descritos por Jampol en el 31,2%, presentaron un cuadro
pseudogripal previo), con una edad entre 10 y 30 años, y con un claro predominio por el
sexo femenino (4/1). El proceso es en general unilateral y agudo, aunque en un 25% se hace
bilateral pero asimétrico (17) y han sido descritos recurrencias en algunos casos (18).
Clínicamente se caracteriza por una disminución rápida de la agudeza visual,
fotopsias y escotomas, a veces con un defecto pupilar aferente relativo y a la
exploración se aprecia la presencia en el fondo de ojo de pequeños focos blanquecinos de
100 a 200 micras de aspecto granuloso, situados a nivel de epitelio pigmentario o
coriocapilar, que tienden a agruparse en media periferia y polo posterior con respeto
foveal. La mácula presenta un aspecto granular anaranjado o blanquecino característica,
diagnóstica en el contexto clínico antes descrito (figs. 6 y 7).
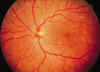 Fig. 6
Fig. 6
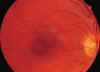 Fig. 7
Fig. 7
Se han descrito otros elementos como la presencia de un edema de papila discreto o
envainamientos venosos; así como un tyndal vítreo en general débil, siendo negativos
los signos inflamatorios del segmento anterior, por lo que en algún caso han sido
erróneamente considerados como un proceso neuroftalmológico (19,20).
En la AFG, los focos blanquecinos, aparecen como puntos hiperfluorescentes desde
tiempos precoces, produciéndose una impregnación tardía por el colorante, que se
extiende desde mácula hasta periferia con un predominio por el polo posterior. Asimismo
puede presentarse una fuga fluoresceínica papilar y más esporádicamente de los
capilares retinianos (fig. 8).
 Fig. 8
Fig. 8
En la ICG, los gruesos vasos coroideos aparecen normales (21), apreciándose en fase
media y tardía del angiograma, múltiples focos hipofluorescentes de 50 a 500 micras
localizados en torno al polo posterior y media periferia, en mucho mayor numero que los
focos visible en la AFG, y que tienden a confluir en el polo posterior, formando a veces
un anillo peripapilar que se correlaciona con el aumento de la mancha ciega descrita en
algunos de estos pacientes, y que desaparece al mismo tiempo que se normaliza la
alteración campimétrica (22,23) (fig. 9).
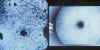 Fig. 9
Fig. 9
En el campo visual, se ha descrito típicamente un aumento de la mancha ciega, aunque
también puede asociarse la presencia de escotoma parafoveales (24), constricción del
campo periférico (25) o incluso escotomas arqueados (26) (fig. 10).
 Fig. 10
Fig. 10
El ERG durante el cuadro agudo, se caracteriza por una alteración especialmente de la
onda a, asociada a una disminución en la amplitud del electrorretinograma precoz ERP y en
menor medida una alteración de la onda b; dicha alteración afecta mas a los bastones y
traduce una evidente disfunción de los fotorreceptores (27) y de los fotopigmentos (28).
El EOG también se encuentra afecto, incluso en casos con poca afectación del ERG (29).
Evolución
Las alteraciones se normalizan en unas semanas (6 a 10), desapareciendo los focos
blanquecinos subretinianos, pudiendo dejar alguna vez, pequeñas alteraciones atróficas
aisladas del epitelio pigmentario retiniano EPR. La agudeza visual se normaliza a valores
superiores a 5/10, normalizándose el ERG y el CV, aunque de manera mucho más lenta que
la agudeza visual.
La presencia de recurrencias es muy rara con algún caso con múltiples recurrencias
bilaterales (30).
Se han descrito como complicaciones excepcionales, la aparición de membranas
neovasculares coroideas, tanto a nivel macular (31) como peripapilar (32).
Etiología
La etiología es desconocida, aunque se han descrito cuadros pseudogripales previos al
cuadro oftalmológico (33-35), asimismo tras una inmunización con vacuna anti hepatitis
B, habiéndose asociado al empleo de dicha vacuna otros cuadros con alteración
neurológica como neuritis óptica y síndrome de Guillen Barré (36). Se ha descrito una
positividad al HLA-B51, 3,7 veces mayor en los pacientes con MEWDS que en la población
normal, curiosamente dicho antígeno se asocia con mayor frecuencia en la enfermedad de
Bechet (37).
III) AMN-AZOOR
A) AMN
Esta entidad fue descrita por Bos y Deutman en 1975 (38), y se caracteriza por afectar
a adultos jóvenes sanos, con predominio por el sexo femenino. Produce una pérdida de
visión rápida bilateral, con presencia de escotomas centrales o paracentrales.
A la exploración oftalmoscópica se aprecia en la forma típica la presencia de una
imagen en trébol o cuñas de aspecto rojo oscuro situado en la fóvea, con mayor
afectación del sector nasal, que incluso puede confundirse con una hemorragia profunda.
En algunos casos la imagen es menos típica y menos marcada, con parches ovalados o
redondos múltiples de color más sonrosado en región central o paracentral de la
mácula, pudiendo coexistir alguna pequeña hemorragia retiniana. No existen signos
inflamatorios ni en el segmento anterior ni en el vítreo.
En la AFG, se aprecia una ligera hipofluorescencia en la zona de la lesión macular, y
en la electrofisiología se caracteriza por la existencia de una alteración de los ERP,
que puede persistir durante meses (39).
En la evolución se produce una lenta resolución espontánea de los síntomas, a lo
largo de semanas o meses.
Su fisiopatología es discutida, aunque para Bos y Deutman se debería a una
alteración de la retina interna, sin embargo los datos electrofisiológicos traducen una
disfunción de los artículos externos de los fotoreceptores (40). Para Sieving (41), el
reflejo más oscuro de la retina a nivel de la lesión se debería a un abombamiento
posterior de la limitante interna por adelgazamiento local de la retina, que hace que la
luz reflejada sobre dicha limitante no llegue hasta el observador (signo de depresión
retiniana de Goldbaum).
Su etiopatogenia es desconocida, sin embargo se ha asociado a proceso pseudogripal, y
se ha descrito tras la inyección de contraste yodado (42) y de epinefrina (43,44), así
como tras el empleo de anticonceptivos (45).
Se plantea la posibilidad de una alteración vascular coroidea o una hipoxia de los
fotorreceptores, sin isquemia visible clínica o angiográfica, producida por la
inyección de contraste o tras el empleo de epinefrina asociado a la situación de shock
hipovolémicos (46,47). La toma de anticonceptivos favorecería dicha hipoxia local. La
otra posibilidad planteada sería la de una interacción entre la epinefrina y la
rodopsina ya que existe una similitud entre la proteína de los receptores adrenérgicos y
esta última (48).
B) AZOOR
En 1993, Gass (49) publica en el J. Clin. Neur. un grupo de 13
pacientes, con un cuadro clínico de pérdida rápida de función en una o varias zonas de
la retina externa traducido bajo forma de escotomas permanentes, con fotopsias y
alteración del ERG siendo el fondo de ojo inicialmente normal. El propio Gass,
considerando a dicho cuadro, así como las otras entidades antes descritas (MEWDS, AMN,
CMF, PIC), formas clínicas diversas de un mismo proceso, caracterizado por la afectación
aguda de la retina externa, decide agruparlos todos bajo la denominación de AZOOR. Sin
embargo, las características tan específicas de los casos publicados por Gass en 1993, y
de los pocos casos recogidos por otros autores hasta la fecha, hace que se deba de
mantener la denominación de AZOOR para los mismos.
Clínica
Esta entidad afecta a mujeres jóvenes en el 70%, de manera uni o bilateral, y se
caracteriza por la aparición de escotomas, asociados a fotopsias (fig. 11).
 Fig. 11
Fig. 11
El fondo de ojo es normal o presenta áreas con mínimas alteraciones pigmentarias a
nivel de EPR y estrechez vascular retiniana más evidentes en la evolución. No existen
síntomas inflamatorios en el segmento anterior, siendo el tyndal vítreo negativo o
débilmente positivo (fig. 12).
 Fig. 12
Fig. 12
La AFG es normal, o presenta un efecto ventana en el área de atrofia del epitelio
pigmentario.
El ERG presenta alteraciones con afectación de la onda a, b así como de los tiempos
implícitos, correlacionándose con la alteración del campo visual. Se han descrito tres
niveles de anomalía (50):
a) El ERG es normal, pero asimétrico con diferencias significativas entre ambos ojos.
b) El ERG está alterado en el ojo afecto.
c) El ERG es patológico en ambos ojos y asimétrico.
En la evolución, las alteraciones campimétricas suelen permanecer con una discreta
reducción de los escotomas, aunque en algunos casos éstos progresan. Solo rara vez se ha
producido una normalización del CV en casos con afectación funcional discreta de la
retina.
Retinopatía aguda externa anular (REAA)
En 1994 Gass (51), describió esta variante del cuadro anterior, en la cual a la
aparición del escotoma, se asociaba en el fondo de ojo un anillo blanquecino de 100-150
micras de grosor rodeando la zona afecta. Dicho anillo desaparecía en 2 semanas, dejando
una placa de atrofia del epitelio pigmentario similar al descrito en los otros casos de
AZOOR (fig. 13).
 Fig. 13
Fig. 13
En la AFG, durante la fase aguda el anillo se comporta como un área hipofluorescente
sin fuga tardía y en fase cicatricial, se produce un efecto ventana en toda la zona de
atrofia del epitelio pigmentario, mientras que la ICG en dicha fase es normal (52) (figs.
14-16).
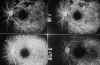 Fig. 14
Fig. 14
 Fig. 15
Fig. 15
 Fig. 16
Fig. 16
Asociaciones
En 1996 Jacobson y cols, presentan un caso de AZOOR, asociado a pleocitosis del LCR y
alteraciones en la resonancia nuclear magnética. Dicho cuadro presenta recurrencias
asociándose una mielitis cervical intermitente, sin datos clínicos de esclerosis
múltiple.
Etiopatogenia
La etiología es desconocida. En 1996 Ara-Iwata y cols (53) encuentran en un paciente
con un cuadro clínico compatible con un AZOOR, una mutación en el gen de la fosfoducina
situado en el cromosoma 1q25-32.1, en el codón 174, en donde una arginina ha sido
cambiado por una lisina. Dicho gen ha sido implicado en algunos procesos degenerativos
retinianos como la enfermedad de Usher.
Tratamiento
No existe tratamiento en la actualidad, no siendo eficaz el empleo de corticoides.
IV) AIBSE
En 1988 Fletcher (54) describió 7 pacientes con un escotoma
temporal agudo por aumento de la mancha ciega, sin alteración oftalmoscópica, con
alteración del ERG y recuperación de la sintomatología en 2 ó 3 meses, definiendo este
nuevo cuadro con la denominación de AIBSE.
Sin embargo, el aumento de mancha ciega ha sido descrita en otros cuadros, como el
MEWDS (55), la CMF (56), AMN (57), POHS (58) y por otra parte, las alteraciones
oftalmoscópicas (manchas blancas) en el MEWDS, se mantienen menos que las alteraciones
campimétricas, por lo que los casos descritos por Fletcher podrían serían una variante
de MEWDS, sin manchas o evolucionado (59).
Todo ello nos hace considerar al AIBSE como un síndrome, caracterizado por el aumento
de mancha ciega sin alteración oftalmoscópica, que puede aparecer en los diversos
cuadros antes descritos y no una entidad independiente. La alteración campimétrica se
debe a una afectación de la retina externa, como lo demuestra por una parte las
características del escotoma con un borde nítido y escarpado, la presencia de fotopsias
en el escotoma, la falta de defectos típicos de la alteración del nervio. y sobre todo
la alteración del ERG (60). Dicha alteración retiniana sería secundaria a la
afectación coroidea peripapilar como lo demuestran los hallazgos en la CM y el MEWDS.
Es importante recordar en estos casos de aumento agudo de la mancha ciega con
normalidad oftalmoscópica, la utilidad del electrorretinograma, para diferenciarlo de los
de causa neurológica.
Palabras clave
Coroiditis multifocal, múltiples puntos blancos evanescentes,
Azoor, neurorretinopatía macular aguda.
Bibliografía
1. Yannuzzi LA, Jampol LM, Rabb M
F y cols: Unilateral acute idiopathic maculopathy. Arch Ophthalmol 1991; 109:
1.411-1.416.
2. Jampol LM, Wiredu A. Editorial,
Mewds, MFC, Pic, AMN, AIBSE, and AZOOR: one disease or many? Retina 1995; 15: 373-378.
3. Khorram KD, Jampol LM, Rosenberg MA: Blind
spot enlargement as a Manifestation of multifocal choroiditis. Arch Ophthalmol 1991;
109: 1.403-1.407.
Hamed LA, Glaser JS, Gass JDM, Schatz NJ: Protacted enlargement of the blind spot in
multiple evanescent white dot syndrome. Arch Ophthalmol 1989; 107: 194-198.
Callanan D, Gass JDM: Multifocal choroiditis and choroidal neovascularization
associated with the multiple evanescent white dot and acute idiopathic blind spot
enlargement syndrome. Ophtahalmology 1992; 99: 1.678-1.685.
Singh. K, de Frank MP, Shults WT, Watzke RC: Acute idiopathic blin spot enlargement -A
spectrum of disease. Ophthalmology 1991; 98: 497-502.
Chittaranjan VR, Brown J, Folk JC, Kimura AE, Gupta S, Walker J: Enlarged blin dpots in
chorioretinal inflmmatory disorders. Ophthalmology 1996; 103: 606-617.
4. Yannuzzi LA, Flower RW, Slakter JS:
Indocyanine green angiography. Mosby 1997; 271-278.
5. Khorram KD, Jampol LM, Rosenberg MA: Blind
spot enlargement as a Manifestation of multifocal choroiditis. Arch Ophthalmol 1991;
109: 1.403-1.407.
7. Spaide RF, Yannuzzi LA, Freund KB: Linear
streaks in multifocal choroiditis and panuveitis. Retina 1991; 11: 229-231.
8. Yannuzzi LA, Flower RW, Slakter JS: Indocyanine
green angiography. Mosby 1997; 276.
9. Spaide RF, Skerry JE, Yannuzzi LA,
DeRosa JT: Lack of the HLA-DR2 specificity in multifocal choroiditis and panuveitis.
Br J Ophthalmol 1990; 74: 536-537.
10. Nolle B, Faul S, Jenisch S, Westphal
E: Periferal multifocal chorioretinitis with panuveitis: clinical and inmunogenetic
characterization in older patients. Graefe´s Arch Clin Exp Ophthalmol 1998; 236:
451-460.
11. Charlotte WTA, Lardenoye MD, Van der
Lelij A, de Loos WS y cols: Peripheral multifocal Chorioretinitis: A distinct clinical
entity? Ophthalmology 1997; 104: 1.820-1.826.
12. Nussenblatt RB, Whitcup SM Palestina
AG: Uveitis: Fundamentals and clinical practice. Mosby St Louis, 1996; 379-381.
13. Kaiser PK, Gragoudas ES: The
subretinal fibrosis and uveitis syndrome. Int Ophthalmol Clin 1996; 36: 145-152.
14. Krill AE,Deutman AF: Acute
retinal pigment epithelitis. Am J Ophthalmol 1972; 74: 193-205.
15. Nolle B, Faul S, Jenisch S, Westphal
E: Periferal multifocal chorioretinitis with panuveitis: clinical and inmunogenetic
characterization in older patients. Graefe´s Arch Clin Exp Ophthalmol 1998; 236:
451-460.
16. Olsen TW, Capone A, Sternberg P,
Grossniklaus HE, Marin DF, Aaberg TM: Subfoveal choroidal neovascularization in
punctate inner choroidopathy.Surgical management and pathologic findings.
Ophthalmology 1996; 103: 2.061-2.069.
17. Aaberg TM, Campo RV,Joffé L: Recurrences
and bilaterality in the multiple evanescent white-dot syndrome. Amer J Ophthalmol
1985; 100: 29-37.
18. Tsai L, Jampol LM, Pollock SC, Olk
J: Chronic recurrent multiple evanescent white dot syndrome. Retina 1994; 14:
160-163.
19. Dodwell DG, Jampol LM, Rosemberg M,
Berman A, Zaret CR: Optic nerve involvement associated with the multiple evanescent
white-dot syndrome. Ophthalmology 1990; 97: 862-868.
21. Darmakusuma I, Glaser BM, Murphy RP,
Gordon LW, Sjaarda RN, Thompson JT: Indocyanine green angiography in multiple
evanescent white-dot syndrome. Amer J Ophthalmol 1994; 117: 7-12.
22. Borruat F, Auger C, Piguet B: Choroidopathy
in multiple evanescent white-dot syndrome. Arch Ophthalmol 1995; 113: 1.569-1.571.
23. Obana A, Masayo Kusumi, Miki T: Indocyanine
green angiographic aspects of multiple evanescent white dot syndrome. Retina 1996; 16:
97-104.
24. Slusher MM, Weaver RG: Multiple
evanescent white dot syndrome. Retina 1988; 8: 132-135.
25. Takeda M, Kimura S, Tamiya M: Acute
disseminated retinal pigment epitheliopathy Folia. Ophthalmol Jpn 1984; 35:
2.613-2.620.
26. Aaberg TM, Campo RV, Joffé L: Recurrences
and bilaterality in the multiple evanescent white-dot syndrome. Amer J Ophthalmol
1985; 100: 29-37.
27. Sieving PA, Fishman GA, Jampol LM,
Pugh: D- Multiple evanescent white dot syndrome II Electrophysiology of the receptors
during retinal pigment epithelial disease. Arch Ophthalmol 1984; 102: 675-679.
28. Van Meel G, Keunen JE, Norren DV,
Kraats JV: Scanning laser densitometry in multiple evanescent white dot syndrome.
Retina 1993; 13: 29-35.
29. Aaberg TM, Campo RV, Joffé L: Recurrences
and bilaterality in the multiple evanescent white-dot syndrome. Amer J Ophthalmol
1985; 100: 29-37.
30. Tsai L, Jampol LM, Pollock SC, Olk
J: Chronic recurrent multiple evanescent white dot syndrome. Retina 1994; 14:
160-163.
31. Wyhinny GJ, Jackson JL, Jampol LM,
Caro NC: Subretinal neovascularization following multiple evanescent white-dot
syndrome. Arch Ophthalmol 1990; 108: 1.384.
32. McCollum CJ, Kimble JA: Perpapillary
subretinal neovascularization associated with multiple evanescent white-dot syndrome.
Arch Ophthalmol 1992; 110: 13-15.
33. Jampol LM, Sieving PA, Puch D,
Fishman GA, Gilbert H: Multiple evanescent white dot syndrome: clinical findings.
Arch Ophthalmol 1984; 102: 671-674.
34. Leys A, Leys M, Jonckheere P, de
Laey JJ: Multiple evanescent white-dot syndrome (mewds). Bull soc belge ophthalmol
1990; 236: 97-108.
35. Laatikainen L, Immonen I: Multiple
evanescent white-dot syndrome. Graefes Arch Clin Exp Ophthalmol 1988; 226: 37-40.
36. Baglivo E, Safran A, Borruat F: Multiple
evanescent white-dot syndrome after hepatitis B vaccine. Amer J Ophthalmol 1996; 122:
431-432.
37. Desarnaulds AB, Borruat FX,Herbort
CP, Spertini F: Le multiple evanescent white dot syndrome: une predisposition
génétique? Klin Mbl Aug 1996; 208-301,302.
38. Bos PJM, Deutman AF: Acute
macular neuroretinopathy. Am J Ophthalmol 1975; 80: 573-584.
39. Sieving PA, Fishman GA, Salzano T,
Rabb MF: Acute macular neuroretinopathy: early recptor potential change suggest
photoreceptor pathology. British J Ophthalmol 1984; 68: 229-234.
40. Priluck IA, Buettner H, Robertson D:
Acute macular neuroretinopathy. Am J Ophthalmol 1978; 86: 775-778.
41. Sieving PA, Fishman GA, Salzano T,
Rabb MF: Acute macular neuroretinopathy: early recptor potential change suggest
photoreceptor pathology. British J Ophthalmol 1984; 68: 229-234.
42. Guzack SV y cols: Acute macular
neuroretinopathy following adverse reactions to intravenous contrast media Retina 1983; 3:
312-317.
43. O'Brien DM, Farmer SG, Kalina RE,
Leon JA: Acute macular neuroretinopathy following intravenous synpathomimetics.
Retina 1989; 9: 281-286.
44. Leys M, Van Slycken S, Koller J, Van
de Somple W: Acute macular neuroretinopathy after shock. Bull Soc Belge Ophthalmol
1991; 241: 95-104.
45. Rush JA y cols: Acute macular
neuroretinopathy. Am J Ophthalmol 1977; 83: 490-494.
46. O'Brien DM, Farmer SG, Kalina RE,
Leon JA: Acute macular neuroretinopathy following intravenous synpathomimetics.
Retina 1989; 9: 281-286.
47. Leys M, Van Slycken S, Koller J, Van
de Somple W: Acute macular neuroretinopathy after shock. Bull Soc Belge Ophthalmol
1991; 241: 95-104.
48. O'Brien DM, Farmer SG, Kalina RE,
Leon JA: Acute macular neuroretinopathy following intravenous synpathomimetics.
Retina 1989; 9: 281-286.
49. Gass JDM: Acute zonal occult
outer retinopathy. J Clin Neuro-ophthalmology 1993; 13: 79-97.
50. Jacobson SG, Morales DS, Sun XK,
Feuer WJ, Cideciyan AV, Gass JD, Milan AH: Pattern of retinal dysfunction in acute
zonal occult outer reinopathy. Ophthalmology 1995: 102: 1.187-1.198.
51. Gass JD, Stern C: Acute annular
outer retinopathy as a variant of acute zonal occult outer retinopathy. Amer J
Ophthalmol 1994; 119: 330-334.
52. Clement Fdez F: Retinopatía
externa zonal aguda. Comunicación presentada a la XII reunión nacional del GEMU.
Madrid 18 febrero 1999.
53. Ara-Iwata F: Analysis of
phosducin as a candidate gene for retinopathies. Ophthalmic Genet 1996 Mar; 17: 3-14.
54. Fletcher WA, Imes RK, Goodman D;
Hoyt WF: Acute idiopathic blind spot enlargement -A big blind spot syndrome without
Optic Disc edema. Arch Ophthalmol 1988; 106: 44-49.
55. Hamed LA,Glaser JS,Gass JDM, Schatz
NJ: Protacted enlargement of the blind spot in multiple evanescent white dot syndrome.
Arch Ophthalmol 1989; 107: 194-198.
56. Khorram KD, Jampol LM, Rosenberg MA:
Blind spot enlargement as a Manifestation of multifocal choroiditis. Arch
Ophthalmol 1991; 109: 1.403-1.407.
57. Singh K, de Frank MP, Shults WT,
Watzke RC: Acute idiopathic blin spot enlargement -A spectrum of disease.
Ophthalmology 1991; 98: 497-502.
58. Callanan D, Gass JDM: Multifocal
choroiditis and choroidal neovascularization associated with the multiple evanescent white
dot and acute idiopathic blind spot enlargement syndrome. Ophtahalmology 1992; 99:
1.678-1.685.
59. Hamed LA,Glaser JS,Gass JDM, Schatz
NJ: Protacted enlargement of the blind spot in multiple evanescent white dot syndrome.
Arch Ophthalmol 1989; 107: 194-198.
60. Chittaranjan VR, Brown J, Folk
JC,Kimura AE, Gupta S, Walker J: Enlarged blind spots in chorioretinal inflmmatory
disorders. Ophthalmology 1996; 103: 606-617.