 Fig. 1
Fig. 1Dres. Rodríguez Galietero A1, Ramón-Capilla M2, Maldonado M1, Cortina P1, Navea A3
Hospital Universitario «La Fe» de
Valencia.
(1) Médico Interno Residente de Oftalmología.
(2) Médico Interno Residente de Neumología.
(3) Médico Adjunto de Oftalmología.
Introducción
La 1.ª descripción de un caso de neurofibromatosis se debe a Von Recklinghausen (1) en 1882. Han existido muchas clasificaciones sobre el tema, englobándose todas las neurofibromatosis bajo el término general de enfermedad de Von Recklinghausen. Sin embargo, recientes descubrimientos de Biología Molecular muestran que existen al menos 2 formas genéticas diferentes, ligadas ambas a una transmisión genética autosómica dominante con alto grado de penetrancia (2):
Neurofibromatosis tipo 1 o enf. de Von Recklinghausen. Es la forma descrita hace años como «clásica o periférica», presentando una incidencia de 1/4.000. Se transmite de forma autosómica dominante y se produce por una mutación genética en el brazo largo del CROMOSOMA 17 (3).
Neurofibromatosis tipo 2. Es la forma descrita hace años como «Central o Acústica Bilateral», y presenta una incidencia de 1/50.000. Su transmisión también es autosómica dominante por una mutación genética producida en el brazo largo del CROMOSOMA 22 (3).
Existen manifestaciones oculares asociadas con cada tipo de neurofibromatosis; así, la Neurofibromatosis tipo 1 se asocia especialmente con nódulos de Lisch, neurofibromas del cuerpo ciliar y coroides, gliomas del nervio óptico, neuromas plexiformes de los párpados; mientras que en la neurofibromatosis tipo 2 se han asociado opacidades subcapsulares posteriores (2).
La enfermedad de Von Recklinghausen está asociada en un 12% a neoplasias (4) y se admite que las células derivadas de la cresta neural tendrían una predisposición especial a la transformación maligna (schwanomas malignos que son los tumores más frecuentes, feocromocitomas y melanomas de la uvea). Los criterios diagnósticos para cada una de las entidades se enunciaron en la «Neurofibromatosis Conference Statement» (5).
Casos clínicos
Caso 1
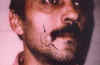
Paciente de 31 años diagnosticado a los 28 de neurofibromatosis tipo 1 (sin antecedentes familiares) tras referir crecimiento de una tumoración peribucal indolora de evolución lenta que tras ser extirpada fue informada con el diagnóstico de neurofibroma plexiforme, que posteriormente recidivó (fig. 1). Tras ello se realizó exploración general exhaustiva en busca de lesiones asociadas encontrando 48 manchas «café au lait» de tamaño superior a 1,5 cm (fig. 2) distribuidas sobre todo en tronco y espalda, neurofibromas en ambos brazos (fig. 3), múltiples Nódulos de Lisch bilaterales. A nivel cerebral y orbitario no se halló patología valorable. Asociaba manifestaciones sistémicas pulmonares (figs. 4, 5 y 6) que consistieron en múltiples lesiones parenquimatosas enfisematosas bullosas más numerosas en vértices pulmonares, que se etiquetaron de neumonía bullosa residual secundaria a su enfermedad general; e imagen redondeada parenquimatosa que se informó como neurofibroma pulmonar.
Fig. 1
 Fig. 2
Fig. 2
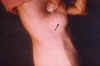 Fig. 3
Fig. 3
Nos fue remitido por pérdida de visión en ojo derecho de 2 meses de evolución con PIO de 2 mmHg, aspecto de «ptisis bulbi» y visión de percepción luminosa. Tras realizar funduscopia y ecografía ocular evidenciamos desprendimiento de retina, sin ningún factor de riesgo asociado para desencadenarlo.
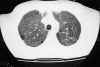 Fig. 4
Fig. 4
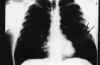 Fig. 5
Fig. 5
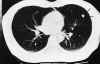 Fig. 6
Fig. 6
Caso 2
Niña de 13 años de edad con padre afecto de neurofibromatosis tipo 1 que a los 5 años de edad fue vista en nuestro servicio por atrofia óptica izquierda. Tras practicarse TAC cerebral se evidenció glioma del nervio óptico izquierdo (fig. 7), sin afectación del nervio óptico derecho. Dado el antecedente familiar se realizó exploración general evidenciándose múltiples manchas «café au lait» y 4 nódulos de Lisch en ambos ojos, etiquetándose el cuadro de neurofibromatosis tipo 1. No se halló patología alguna a nivel sistémico. En estudio tomográfico realizado 2 años después destacaba la desaparición de la lesión gliomatosa del nervio óptico sin presencia de nuevas lesiones acompañantes. En controles sucesivos hasta el año 90 la niña estuvo estable; en enero de 1991 presentó focalidad neurológica y en las imágenes tomográficas se apreciaron lesiones gliomatosas a nivel parietal derecho, hemisferio cerebeloso derecho, mesencéfalo (afectando la zona vecina al acueducto de Silvio) produciendo hidrocefalia (de la que fue intervenida mediante la colocación de una válvula de derivación ventrículo-peritoneal). En controles posteriores las lesiones gliomatosas del parénquima cerebral aumentaron de tamaño a pesar del tratamiento radioterápico recibido y las lesiones del nervio óptico no han regresado (figs. 8, 9, 10, 11, 12, 13 y 14).
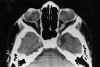 Fig. 7
Fig. 7
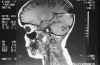 Fig. 8
Fig. 8
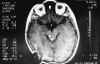 Fig. 9
Fig. 9
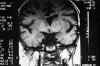 Fig. 10
Fig. 10
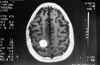 Fig. 11
Fig. 11
 Fig. 12
Fig. 12
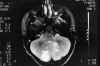 Fig. 13
Fig. 13
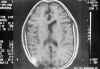 Fig. 14
Fig. 14
Discusión
Existen manifestaciones oculares frecuentemente descritas en el curso de la neurofibromatosis tipo 1:
Nódulos de Lisch. Son hamartomas melanóticos del iris, que forman elevaciones sobre la superficie de éste de color amarillento o castaño, por lo que hay que hacer diagnóstico diferencial con los Nevus de iris. Se piensa que son específicos de la Neurofibromatosis tipo 1, presentándose de forma bilateral (6) (como el caso 1) constituyendo el hallazgo ocular más frecuente (6,7), siendo su detección vital para investigar tumores asociados y realizar consejo genético (8). Se han descrito de forma ocasional en la Neurofibromatosis tipo 2 (9), donde presentan típicamente un patrón unilateral, en la enfermedad de Cushing (10) (adenoma hipofisario basófilo) y algún tipo de MEN (11) (Neoplasia Endocrina Múltiple).
La prevalencia de estos nódulos aumenta con la edad en los pacientes con neurofibromatosis, presentándolos el 50% de pacientes con 5 años de edad, el 75% de pacientes con 15 años de edad y el 95% de pacientes con 30 años de edad (8) (probablemente el número de ellos esté ligado al momento del diagnóstico; así en el caso 1 existían múltiples nódulos bilaterales al diagnóstico, que se realizó a los 28 años y en el caso 2 sólo 4 nódulos en cada ojo, diagnosticándose a los 5 años.
Displasias o agenesia del esfenoides. Sobre todo aparecen a nivel del ala mayor produciendo un exoftalmos pulsátil.
Neurofibromas de cuerpo ciliar y coroides.
Gliomas del nervio óptico. En ellos hay que tener en cuenta su posible regresión espontánea (como el caso 2). Esta regresión ha sido descrita en la literatura de forma ocasional (12). En nuestro caso no se extirpó ni se irradió controlándolo periódicamente porque estos gliomas son hamartomas astrocíticos benignos y éste ya había producido atrofia óptica importante y normalmente tras un período de crecimiento dejan de aumentar, aunque en muy pocos casos regresan. En el niño la alteración funduscópica más frecuente es la «Atrofia "ptica» o «Edema Papilar» secundarios al glioma del nervio óptico.
Neuromas plexiformes de los párpados. Se asocian muchas veces a glaucomas, aunque la patogenia de estos glaucomas no está aclarada, postulándose teorías al respecto: a) tejido neurofibromatoso en el ángulo; b) anomalía de desarrollo del ángulo; c) cierre del ángulo por un tumor del cuerpo ciliar.
Se han descrito en la literatura algunas alteraciones oculares asociadas a la neurofibromatosis tipo 2 entre ellas: tumores intracraneales y espinales (13), opacidades cristalinianas subcapsulares posteriores (14), hamartomas retinianos (15,16) y del epitelio pigmentario asociados y no asociados a membranas epirretinianas», meningiomas bilaterales del nervio óptico (18). Cualquier manifestación para que ser incluida en el diagnóstico de neurofibromatosis tipo 2 debe presentar una serie de criterios diagnósticos (5):
1) Neurinomas Acústicos Bilalerales o ...
2) Riesgo de primer grado de sufrir neurofibromatosis tipo 11 con neurinoma acústico unilateral o ...
3) Dos de los siguientes: neurofibroma, meningioma, glioma, schwannoma, opacidad cristaliniana subcapsular posterior.
El signo clínico más importante son las manchas «café au lait» que aunque típicas no son patognomónicas. Deben encontrarse en número superior a 6 y de tamaño mayor de 1,5 cm (19). Se encuentran en más del 99% de enfermos y aparecen sobre todo en las áreas no expuestas. Nuestros dos pacientes presentaban múltiples manchas especialmente en el tronco.
Los neurofibromas se pueden desarrollar tanto en el sistema nervioso central como en el periférico y su clínica dependerá de la localización. En el caso 2 la enferma presentó cefalea secundaria a hidrocefalia y sintomatología de lóbulo parietal debido a la localización de un glioma ahí, así como síndrome cerebeloso destacando inicialmente ataxia para en controles sucesivos presentar un síndrome cerebeloso de ataxia-asbasia.
La posibilidad de afectación visceral es variada, siendo el tracto gastrointestinal el sistema más afecto, apareciendo múltiples neurofibromas. También se ha descrito la afectación de hígado, pulmón (20), riñón, vejiga y corazón. A nivel pulmonar las lesiones son muy infrecuentes; entre ellas: fibrosis difusa intersticial (21), múltiples enfisemas bullosos (22), neumonía descamativa bullosa y neurilemas torácicos (23). Nuestro caso 1 presentaba neurofibroma de parénquima pulmonar diagnosticado por biopsia e imágenes bullosas enfisematosas múltiples calificadas como secundarias al proceso general, siendo ambas manifestaciones muy poco frecuentes en el curso habitual de la enfermedad.
Resumen
La Neurofibromatosis es una facomatosis que se transmite de forma autosómica dominante con penetrancia variable, pudiendo acompañar a síndromes de neoplasias endocrinas múltiples (MEN). Algunos pacientes presentan manifestaciones al nacimiento y más del 60% de los afectados son diagnosticados antes de los 2 años de edad.
Clínicamente se combinan manchas «café au lait» de más de 1,5 mm de tamaño y en número mayor de 6 con tumoraciones de las vainas de las células de Schwann (neurofibromas). En general (aunque depende del tipo de neurofibromatosis de la que se trate) los hallazgos oftalmológicos se concretan en Nódulos de Lisch en el iris, neurofibromas palpebrales, tumores intraorbitarios, glaucomas congénitos, glioma óptico, etc.
Presentamos dos casos excepcionales de esta enfermedad. El primero se diagnosticó a los 28 años de edad, y presenta aparte de las manifestaciones típicas, múltiples nódulos de Lisch bilaterales, desprendimiento de retina espontáneo, neurofibroma de parénquima pulmonar y múltiples lesiones enfisematosas bullosas pulmonares (manifestaciones estas tres últimas muy infrecuentes). El segundo caso debutó con glioma del nervio óptico que produjo atrofia óptica, que regresó espontáneamente sin tratamiento, así como gliomas parietales, cerebeloso y mesencefálico que originaron hidrocefalia e hipertensión endocraneal.
Bibliografía