 Fig. 1
Fig. 1Dres. Gómez de Liaño Sánchez R1, Alcocer Alfonso M.ªP2, Ortega Usobiaga J3
(1) Doctor en Medicina. Instituto de
Investigaciones Oftalmológicas Ramón Castroviejo. Madrid.
(2) Licenciado en Medicina. Valencia.
(3) Licenciado en Medicina. Instituto de Investigaciones Oftalmológicas Ramón
Castroviejo. Madrid.
Introducción
El término oftalmoplejía crónica externa progresiva (OCEP), tal como se utiliza habitualmente, incluye un grupo de entidades diferentes que tienen en común el desarrollo lento y progresivo de una oftalmoplejía externa general. La presencia de una OCEP sugiere una relación con una citopatía mitocondrial, pero no todos los pacientes tienen esta alteración (1,2).
Caso clínico
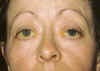
Mujer de 41 años de edad que acude a la consulta de Estrabología por presentar un cuadro de limitación de la motilidad ocular y blefaroptosis. Entre sus antecedentes destaca un estrabismo adquirido a los 2 años de edad y operado a los 37; la cirugía practicada había sido una retroinserción y cirugía del hilo en el recto medial de ambos ojos. En la exploración, la paciente presentaba:
a) Agudeza visual (lejos):
OD (+1,75+1,25x80°) = 20/20;
OI (+1,50+2,75x95°) = 20/20.
b) Agudeza visual (cerca): 20/20 en AO.
c) Motilidad ocular externa:
d) Binocularidad: correspondencia retiniana anómala con supresión del OI.
e) Blefaroptosis del OI de 3 mm, pero con buena función del elevador.
f) Resto de la exploración oftalmológica: normal.
A lo largo de los 5 años siguientes se produjo un aumento progresivo de la limitación de todos los movimientos oculares, así como de la blefaroptosis. Durante este tiempo los estudios neurológicos fueron normales, pero al cabo de estos 5 años acabó confirmándose la patología sistémica. Se encontró un título de anticuerpos antimicrosomales de 1/6400.
Fig. 1
![]() Fig. 2
Fig. 2
Discusión
Existen diferentes cuadros de OCEP:
a) Síndrome de Kearns-Sayre. Afecta a la primera infancia. Se describió inicialmente como una combinación de retinitis pigmentaria, oftalmoplejía externa y bloqueo de la conducción cardíaca. Para el diagnóstico, actualmente, se exige la tríada compuesta por retinitis pigmentaria, oftalmoplejía e inicio antes de los 20 años de edad; además, debe existir una de las siguientes situaciones: proteinorraquia de más de 100 mg/dl, bloqueo de la conducción cardíaca y síndrome cerebeloso. Puede asociarse a otras alteraciones, como pérdida de audición, debilidad de las extremidades, diabetes mellitus, hipoparatiroidismo, irregularidades menstruales, déficit de hormona del crecimiento y disfunción tubular renal (generalmente síndrome de Debré - De Toni - Fanconi); ocasionalmente aparecen episodios paralíticos (3). La mayoría de los casos son esporádicos. El ADN mitocondrial puede sufrir delecciones y duplicaciones (4). Es posible que una zona del ADN deleccionado se duplique más rápidamente, ya que se ha encontrado una acumulación de ADN mitocondrial con el paso de los años (5-7).
b) OCEP aislada. Aparece en la infancia y la adolescencia. Es un cuadro menos grave. Se caracteriza por una OCEP con blefaroptosis lentamente progresiva. El paciente no suele referir diplopía. Se asocia frecuentemente a debilidad de las extremidades. La mayoría son esporádicos, aunque se ha encontrado dos formas con herencia autosómica dominante ligadas a los cromosomas 10q y 3p, así como una con herencia autosómica recesiva (1,7).
c) OCEP «plus». Es el diagnóstico que dimos a nuestra paciente. Presenta el mismo fenotipo que el síndrome de Kearn-Sayre pero no cumple todos los criterios para su diagnóstico. No se ha podido clasificar, tampoco, como una alteración bioquímica o mitocondrial, aunque es posible que haya un cierto aumento de DNA mitocondrial. Existe una forma rara de OCEP autosómica dominante que se inicia al final del tercer decenio de vida y cursa con blefaroptosis, disfonía, diafagia, debilidad facial y de las extremidades, cataratas, intolerancia al ejercicio y vida media de 40-50 años. En estas familias se han encontrado delecciones múltiples del DNA mitocondrial (frente a la delección única del Kearn-Sayre), cuyo tamaño y localización no influyen en la gravedad del cuadro. En las fibras musculares se han encontrado fibras rojas rasgadas (red ragged fibres) con deplección de citocromo-oxidasa y acumulación de mitocondrias anormales (2,6,9).
d) Otros cuadros asociados a OCEP: síndrome de Refsum, ataxia de Sanger-Brown, ataxia familiar amiotrófica neural, encefalopatía espongiforme, distrofia oculofaríngea, tetraplejia espástica con retinitis pigmentaria, distrofia muscular con miopatía y miotonía ocular y distal, oftalmoplejía externa amiotrófica, y otros procesos menos frecuentes.
En la OCEP se halla afectada la cadena respiratoria mitocondrial. Como consecuencia, disminuye la síntesis de ATP y se acumulan catabolitos y lactato. Para mantener la producción de ATP aparece una compensación lipídica.
El consejo genético es difícil. Una vez instaurada la clínica se ha propuesto, en niños, un régimen cetogénico hiperlipídico e instaurar un seguimiento estrecho, para detectar la extensión multivisceral y las complicaciones del régimen hiperlipídico.
La debilidad muscular es simétrica. La motilidad ocular intrínseca es normal. El diagnóstico, sobre todo inicialmente, es difícil, ya que los signos electromiográficos son de difícil valoración y la biopsia (de orbicular o músculo extraocular) no siempre arroja resultados concluyentes.
Resumen
Se presenta el caso de una paciente adulta que presenta una oftalmoplejía crónica externa progresiva, asociada a blefaroptosis. La paciente había sido intervenida quirúrgicamente de esotropía esencial cuatro años antes. Tras un estudio oftalmológico y sistémico, y un seguimiento de cinco años, se catalogó como oftalmoplejía crónica externa progresiva «plus». El diagnóstico fue difícil debido a la infrecuencia del proceso, la similitud con otros cuadros y la dificultad de la confirmación sistémica. Se describe su evolución durante cinco años.
Palabras clave
Oftalmoplejía crónica externa progresiva, síndrome de Kearns-Sayre, citopatía mitocondrial.
Summary
We report a case of an adult woman presenting a chronic progressive external ophthalmoplegia, associated with blepharoptosis. The patient had been surgically treated because of an essential esotropia four years before. After an ophthalmic and general examination, and a five years follow-up, a chronic progressive external ophthalmolplegia «plus» was diagnosed. The diagnosis was difficult due to the infrequency of the process, its similarity with other diseases and the difficulty of a systemic confirmation. A five years follow-up is described.
Key words
Chronic progressive external ophthalmoplegia, Kearns-Sayre syndrome, mitochondrial cytopathy.
Bibliografía