Fig. 1. Retinografía del ojo derecho e izquierdo del paciente.
SEMINARIO DE CASOS CLÍNICOS
ÁBALO LOJO JM1, BARREIRO RODRÍGUEZ L1, GÓMEZ-ULLA DE IRAZAZÁBAL FJ2
(1) Licenciado en Medicina y Cirugía. Médico
Interno Residente de Oftalmología. Complejo Hospitalario Universitario de
Santiago de Compostela (CHUS).
(2)
Catedrático de Oftalmología. Universidad de Santiago de Compostela. Jefe de
Sección de la Unidad de Retina Médica y Diabetes Ocular (CHUS).
La epiteliopatía pigmentaria placoide multifocal posterior aguda (EPPPMA) es una rara enfermedad idiopática del epitelio pigmentario de la retina. Afecta fundamentalmente a personas jóvenes presentándose como una disminución rápida de la visión central bilateral y asimétrica con afectación del ojo adelfo en pocos días siendo el caso descrito una excepción. Oftalmoscópicamente, lo más característico es la aparición de lesiones placoides, multifocales, blanco-amarillentas, menores de un disco de diámetro y localizadas en el polo posterior a nivel del epitelio pigmentario de la retina. La evolución es generalmente benigna y las lesiones se resuelven en 2-4 semanas dejando alteraciones residuales del epitelio pigmentario.
CASO CLÍNICO
Mujer de 39 años, sana sin ningún antecedente de interés. Acudió en mayo de 2002 remitida desde otro centro por disminución de agudeza visual y miodesopsias en el ojo derecho de 10 días de evolución, a tratamiento con 25 mg de prednisona al día.
La agudeza visual con corrección era de 20/80 en el ojo derecho y de 20/20 en el ojo izquierdo. La exploración del segmento anterior y presión intraocular fue normal en ambos ojos. Oftalmoscópicamente en el ojo derecho se objetivaron múltiples lesiones placoides coalescentes blancoamarillentas, de tamaño entre un cuarto y un diámetro papilar, situado por dentro de las arcadas vasculares, afectando fundamentalmente el polo posterior y media periferia. El ojo izquierdo no presentaba alteraciones significativas. Se decide en ese momento poner una pauta de supresión progresiva de corticoides.
![]()
Fig. 1. Retinografía del ojo derecho e izquierdo
del paciente.
Acude al mes objetivándose en la exploración una mejoría de la agudeza visual 20/25 en el ojo derecho y 20/20 en el ojo izquierdo. El fondo del ojo derecho presentaba lesiones de color blanco grisáceo coalescentes con zonas despigmentadas o con acúmulo de pigmento con aspecto de inactividad y otras con una coloración amarillenta cremosa que daban la impresión de estar activas. Se le realizó AGF apreciándose lesiones en fase aguda con una hipofluorescencia temprana y posterior hiperfluorescencia difusa y lesiones en estadio cicatricial que mostraban una hiperfluorescencia precoz por un efecto ventana.
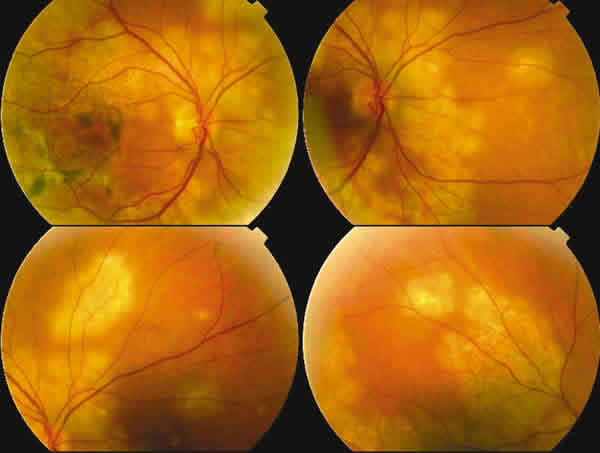
Fig. 2. Lesiones activas e inactivas de EPPPMA.
A los 3 meses la paciente presentaba una agudeza visual en el ojo derecho de 20/20 y T1 en visión próxima pero seguía refiriendo discretas metamorfopsias. Continuaban apreciándose placas multifocales de color blancogrisáceo, aparentemente inactivas. A los 4 meses se realizaron retinografías, AGF e ICV demostrando que las lesiones se encontraban en inactividad, manteniéndose a los 7 y 12 meses.
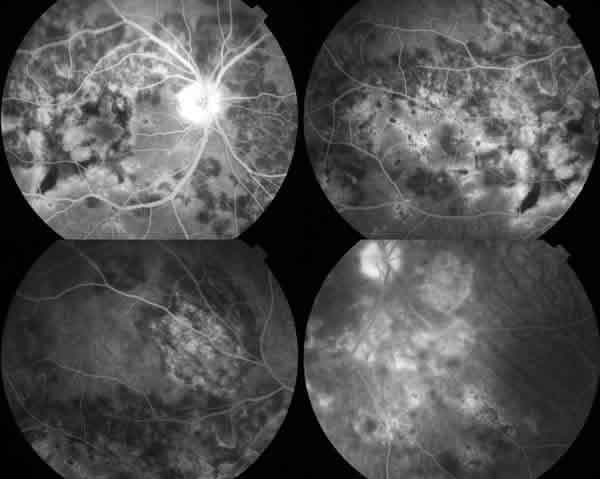
Fig. 3. AGF de lesiones de EPPPMA.
La paciente acudió en agosto de 2004 a la unidad de Urgencias de Oftalmología refiriendo miodesopsias en el ojo izquierdo con una agudeza visual de 20/20 en ambos ojos. En la exploración oftalmoscópica se visualizan lesiones placoides activas en el ojo izquierdo y cicatriciales en el derecho. Se realizó AGF demostrando lesiones en actividad en el ojo izquierdo. Se le instauró tratamiento con prednisona de 30 mg/día.
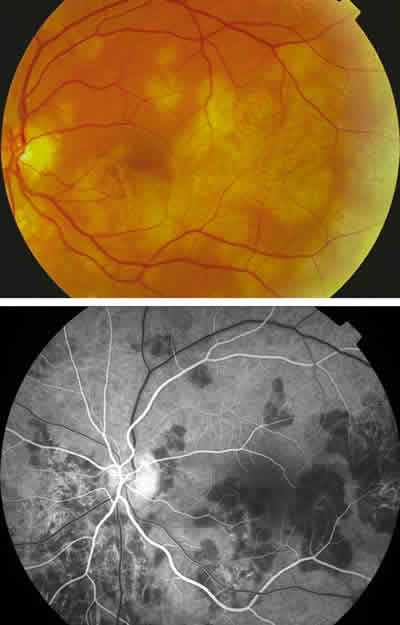
Fig. 4. Retinografía y angiografía del ojo izquierdo.
La paciente acude a los dos meses refiriendo una ligera mejoría pero continuaban la presencia de placas activas en el ojo izquierdo. A los 4 meses del comienzo de la clínica en el ojo izquierdo se le realizó una nueva AGF no mostrando ya signos de actividad y con una agudeza visual de 20/20 en ambos ojos.
DISCUSIÓN
La EPPPMA fue descrita por primera vez por Gass (1) en 1968 . La etiopatogenia de la EPPPMA todavía no ha sido aclarada, pero algunos trabajos sugieren que puede tratarse de un proceso autoinmune que desemboca en una angeítis coroidea precapilar con hipoperfusión coriocapilar y afectación secundaria del epitelio pigmentario (2,3).
Afecta fundamentalmente a personas jóvenes, sanas con edades comprendidas entre los 20 y los 50 años. Aproximadamente un tercio de los pacientes tienen una infección viral previa (1). Se han descrito casos de EPPPMA asociados a granulomatosis de Wegener (4), tuberculosis (5), colitis ulcerosa (6), infección por Streptococos del grupo A (7), vacunación previa de hepatitis B (8), sarcoidosis (9) y enfermedad de Lyme (10), aunque no está clara la naturaleza de esta asociación. También se ha sugerido relación con los antígenos de HLA-B7 y HLA-DR2 (11).
Clínicamente, se presenta como una disminución rápida de la visión central de un ojo asociada con escotomas centrales o paracentrales. La pérdida visual es generalmente bilateral y asimétrica, con afectación del ojo adelfo en pocos días siendo el caso descrito una excepción ya que el ojo adelfo se afecta 2 años más tarde.
Oftalmoscópicamente, lo más característico es la aparición de lesiones placoides, multifocales, blanco-amarillentas, menores de un disco de diámetro y localizadas en el polo posterior a nivel del epitelio pigmentario de la retina.
A partir de la primera semana las lesiones empiezan a palidecer espontáneamente y después de 2 ó 3 semanas esas áreas se reemplazan por alteraciones variables del EPR, como atrofia e hiperpigmentación. Pueden verse hallazgos atípicos como papilitis, periflebitis, oclusión de la vena central de la retina, neovascularización papilar, desprendimiento de retina neurosensorial y la hemorragia subhialoidea (12,13).
La angiografía con fluoresceína (AGF) revela un típico patrón de «bloqueo precoz, tinción tardía». En la fase precoz del angiograma, las lesiones agudas son hipofluorescentes. La hipofluorescencia se debe a la opacificación del EPR y a la falta de perfusión coroidea. Las lesiones se hacen hiperfluorescentes en las fases tardías de la prueba. En las fases más tardías la AGF muestra hiper/hipofluorescencia variables, según la extensión de las alteraciones del EPR. La angiografía con verde de indocianina (AVI) muestra hipofluorescencia de las lesiones activas y cicatrizadas, resaltando la importancia de la falta de perfusión coroidea en la EPPPMA.
La evolución es generalmente benigna y las lesiones se resuelven sin tratamiento en 2-4 semanas dejando alteraciones residuales del epitelio pigmentario. La visión mejorará al hacerlo las lesiones pero no siempre se normalizan por completo pudiendo persistir en algunos pacientes escotomas paracentrales residuales o síntomas de metamorfopsia. Raramente presentan recurrencias. Se ha recomendado el tratamiento con corticoides sistémicos en casos de afectación foveal o asociados a vasculitis del SNC (14).
BIBLIOGRAFÍA
Gass JDM. Acute posterior multifocal placoid pigment epitheliopathy. Arch Ophthalmol 1968; 80: 177-185.
Howe LJ, Woon H, Gram. EM, Fitzke F, Bhandari A, Marshall J. Choroidal hypoperfusion in acute posterior placoid pigment epitheliopathy. Ophthalmology 1995; 102: 790-798.
Park D, Schatz H, McDonald R, Johnson RN. Indocyanine green angiography of acute multifocal posterior placoid pigment epitheliopathy. Ophthalmology 1995; 102: 1877-1883.
Chiquet C, Lumbroso L, Denis P, Papo T, Durieu I, Lehoang P. Acute posterior multifocal placoid pigment epitheliopathy associated with Wegeners granulomatosis. Retina 1999; 19(4): 309-313.
Anderson K, Patel KR, Webb L, Dutton GN. Acute posterior multifocal placoid pigment epitheliopathy associated with pulmonary tuberculosis. Br J Ophthalmol 1996; 80: 186.
Di Crecchio, Parodi MB, Saviano S, Ravalico G. Acute posterior multifocal placoid pigment epitheliopathy and ulcerative colitis: a possible association. Acta Ophthalmol Scand 2001; 79: 319-321.
Lowder CY, Foster RE, Gordon SM, Gutman FA. Acute posterior multifocal placoid pigment epitheliopathy after acute group A streptococcal infection. Am J Ophthalmol 1996; 122: 115-117.
Bourges JL, Pisella PJ, Laurens C, Limon S. Multifocal placoid epitheliopathy and anti-hepatitis B vaccination. J Fr Ophthalmol 1998; 21: 696-700.
Bodiguel E, Benhamou A, Le Hogan P, Gautier JC. Cerebral infarction, placoid epitheliopathy and sarcoidosis. Rev Neurol (Paris) 1992; 148(12): 746-751.
Bodine SR, Marino J, Camisa TJ, Salvate AJ. Multifocal choroiditis with evidence of Lyme disease. Ann Ophthalmol 1992; 24: 169-173.
Wolf MD, Folk JC, Panknen CA, Goeken NE. HLA-B7 and HLA-DR2 antigens and acute posterior multifocal placoid pigment epitheliopathy. Arch Ophthalmol 1990; 108: 698-700.
De Souza S, Aslanides IM, Altomare F. Acute posterior multifocal placoid pigment epitheliopathy associated with retinal vasculitis, neovascularization and subhyaloid hemorrage. Can J Ophthalmol 1999; 34: 343-345.
Abu El-Asrar AM, Aljazairy AH. Acute posterior multifocal placoid pigment epitheliopathy with retinal vasculitis and papillitis. Eye 2002; 16: 642-644.
OHalloran HS, Berger JR, Lee WB et al. Acute multifocal placoid pigment epitheliopathy and central nervous system involvement: nine new cases and review of the literature. Ophthalmol 2001; 108: 861-868.