
Fig. 1. Defectos paracentrales en la campimetría del caso 1.
SEMINARIO DE CASOS CLÍNICOS
SUÁREZ BARAZA J1, GUTIÉRREZ DÍAZ A2
Hospital Universitario 12 de Octubre.
Madrid. España.
(1) Licenciado en Medicina.
(2) Doctor en Medicina.
RESUMEN
Caso Clínico: Presentamos dos casos de distrofias patrón del epitelio pigmentario retiniano. Destacamos el de una joven con flecos retinianos y un déficit de agudeza visual inusual a edades tan tempranas. El segundo representa la evolución típica de esta enfermedad.
Discusión: Las distrofias patrón no deben ser consideradas necesariamente una condición benigna hasta etapas avanzadas de la vida ya que pueden conducir a un déficit visual precoz, siendo necesario el diagnóstico diferencial con otras entidades con las que comparte signos oftalmoscópicos comunes en su evolución.
Palabras Clave: Distrofia patrón del epitelio pigmentario retiniano, Fundus flavimaculatus, Degeneración macular asociada a la edad.
INTRODUCCIÓN
Las distrofias patrón se caracterizan por desarrollar distintos patrones de depósitos pigmentarios subretinianos (amarillentos, naranjas o grisáceos) formando placas o enrejados en el área macular.
Su curso clínico es relativamente benigno, con agudezas visuales preservadas hasta edades avanzadas (1).
CASO CLÍNICO 1
Paciente marroquí de 25 años con déficit de agudeza visual (AV) en ambos ojos (AO).
La AV era de cuenta dedos a un metro en ojo izquierdo (OI) y 0,3 en ojo derecho (OD) con corrección. El segmento anterior era normal.
Se encontraba en tratamiento con timolol 0,5% por glaucoma primario de ángulo abierto, aunque al diagnóstico ya presentaba una atrofia óptica en OI.
En la perimetría computarizada Humphrey 30/2 del OD se observaban tres puntos agrupados en los 15º centrales con una P<0.5% en una campimetría no glaucomatosa (Humphrey instruments, modelo 740, Carl Zeiss Inc) (fig. 1).
Fig. 1. Defectos paracentrales en la campimetría
del caso 1.
El test de visión cromática Farnsworth-Munsell 28-Hue en OD era normal.
En la oftalmoscopía tenía un parénquima retiniano aparentemente normal con algunas zonas grisáceas subretinianas en el polo posterior de AO y en el OD unos flecos amarillentos perimaculares (figs. 2 y 3).
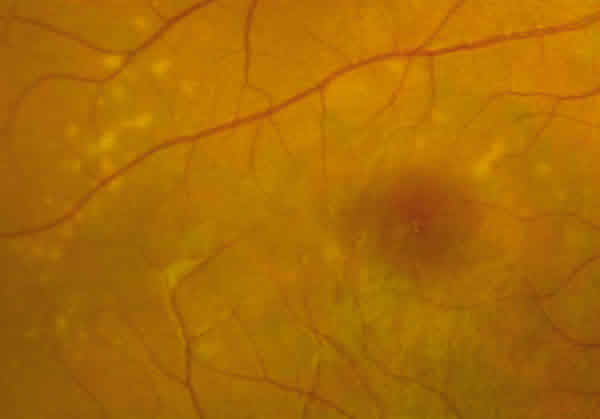
Fig. 2. Detalle de los flecos retinianos perimaculares en OD del caso 1.
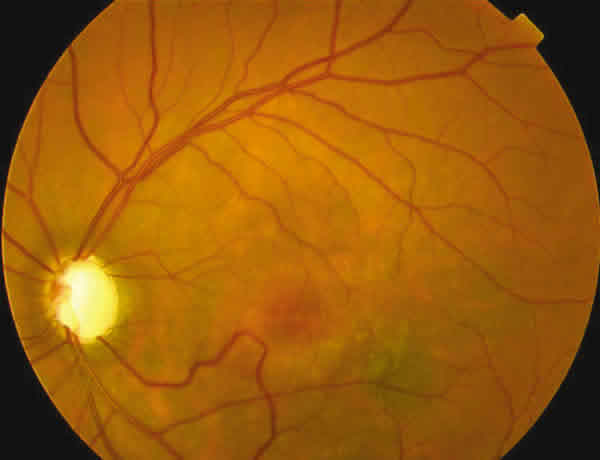
Fig. 3. Retinografía del OI del caso 1
La papila izquierda era pálida y con una excavación de 10/10. En la arcada vascular inferior se apreciaba una malformación vascular tipo anastomosis arteriovenosa. La papila derecha no presentaba un aspecto glaucomatoso.
En la angiografía fluoresceínica (AFG) presentaba un patrón anular perimacular de aumento de la fluorescencia coroidea junto con un patrón reticular con efecto pantalla, siendo los flecos hipofluorescentes (figs. 4 y 5).
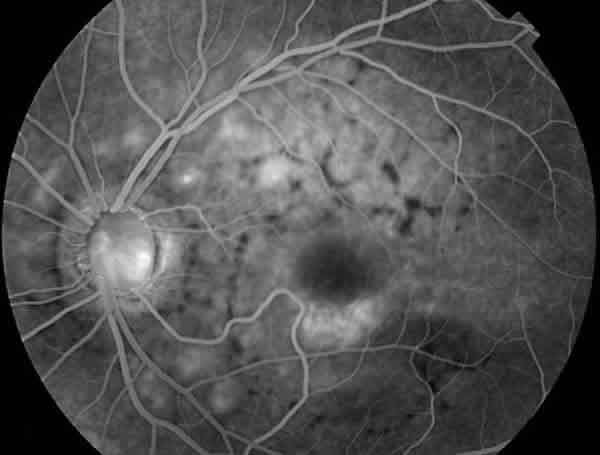
Fig. 4. AFG del OI del caso 1. Aumento de fluorescencia coroidea y patrón
reticular con efecto pantalla.
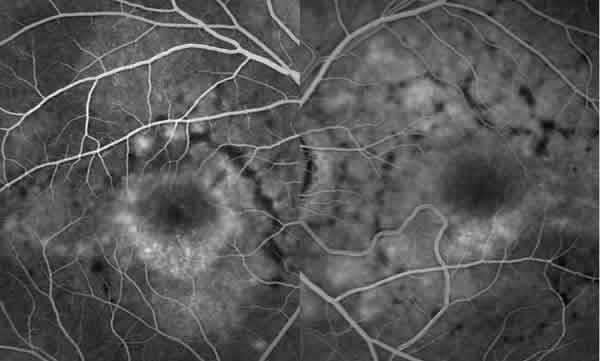
Fig. 5. AFG de AO del caso 1. Detalle del polo posterior de AO.
Las pruebas electrofisiológicas (potenciales evocados y electrorretinograma) fueron normales.
Su madre y hermanos estaban afectados de una enfermedad retiniana que les dificultaba la visión, aunque no fue posible estudiar a la familia por no vivir en España.
Durante tres años de seguimiento no ha habido cambios clínicos significativos.
La clasificamos en el grupo III de Gass de las distrofias patrón (patrón reticular) compartiendo signos del grupo IV (patrón multifocal).
CASO CLÍNICO 2
Paciente de 70 años remitida para cirugía de cataratas.
En la revisión preoperatoria llamaba la atención una atrofia del epitelio pigmentario retiniano (EPR) macular sin depósitos pigmentarios evidentes. Fue considerada entonces como una Degeneración macular asociada a la edad (DMAE) atrófica.
Tras un postoperatorio sin incidencias la AV era de 0,3 AO.
En la AFG presentaba una hiperfluorescencia coroidea macular en el seno de la cual destacaba un patrón radial de líneas hipofluorescentes (figs. 4 y 5). La clasificamos en el grupo II de las distrofias patrón según Gass (patrón tipo mariposa).
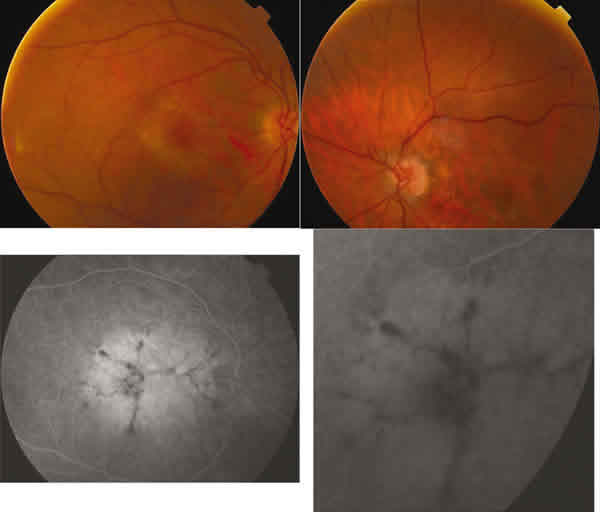
Fig. 6. Retinografías y AFG de AO en caso 2. Depósito pigmentario con patrón en
mariposa.
DISCUSIÓN
Las distrofias patrón fueron clasificadas por JDM Gass en cinco grupos dependiendo del patrón de distribución pigmentaria.
Existe una gran de heterogeneidad clínica en familias con esta condición, resultado de la progresión del fenotipo con la edad. Algunos enfermos comparten combinaciones de signos de los cinco grupos (1).
Nuestra primera paciente pertenece al grupo III (reticular) con un patrón pigmentario perimacular en forma de red mas aparente angiográfica que oftalmoscopicamente. También se observan signos del grupo IV ( patrón multifocal) que simula un Fundus flavimaculatus (FF) con flecos interconectados (1).
Aunque las distrofias patrón son asintomáticas en jóvenes y con cambios oftalmoscópicos difíciles de reconocer, la baja AV de nuestra primera paciente y la presencia de flecos maculares obligan a hacer un diagnóstico diferencial con la enfermedad de Stadgardt- Fundus flavimaculatus. Estos pacientes suelen debutar precozmente con bajas agudezas visuales y sin signos oftalmoscópicos aparentes (disociación clínico-oftalmoscópica).
Algunas distrofias patrón pueden compartir con el FF los flecos perimaculares y la atrofia anular del EPR macular pero la ausencia del silencio coroideo (típico signo angiográfico de la enfermedad de Stadgardt-FF) confirma el diagnóstico de distrofia patrón (1,2). Las pruebas electrofisiológicas pueden ser normales en ambas distrofias.
El patrón hereditario también diferencia ambas entidades (recesivo el FF, dominante la distrofia patrón) excepto la variante reticular que puede heredarse de ambas formas (1).
La principal causa del déficit visual en OI es la atrofia óptica glaucomatosa. No hemos encontrado en la literatura relación entre estas dos enfermedades, aunque si se relaciona con otras como el pseudoxantoma elástico y la distrofia mío tónica (1).
Histopatologicamente se ha visto que los flecos de las distrofias patrón que simulan un Fundus flavimaculatus no son causados por depósitos de lipofucsina en el EPR, sino por productos de degradación de las células del EPR y fotorreceptores (1).
Otro factor común entre estas dos entidades y encontrada en varias familias es la mutación del gen periferina/RDS (retinal degeneration slow) localizado en el brazo corto del cromosoma 6. Mutaciones de este gen también incluyen fenotipos de Retinosis pigmentaria demostrando expresiones variables de un desorden genético común (3-5).
La red pigmentaria puede ser reemplazada en etapas ulteriores por cambios atróficos del epitelio pigmentario, de hecho se han descrito casos de degeneración macular geográfica en la evolución de las distrofias patrón, siendo catalogados por algunos como una manifestación tardía de las mismas. No se puede afirmar si la atrofia del EPR es el resultado directo de la distrofia o la distrofia predispone a una temprana y mayor susceptibilidad a la DMAE (6).
Nuestro segundo caso representa la evolución habitual de esta enfermedad, una paciente de 70 años con un progresivo pero lento déficit de AV, confundido en un principio con una DMAE.
En un seguimiento a largo plazo de pacientes con distrofias patrón la mayoría mantenían visiones estables hasta los setenta años pero algunos desarrollaron bajas AV debido a escotomas paracentrales correspondientes a zonas de pérdida de la coriocapilar perifoveal (7). En nuestro primer caso podemos encontrar estos escotomas (fig. 1).
Las distrofias patrón del EPR no deben ser consideradas necesariamente una condición benigna ya que pueden conducir a un déficit visual precoz siendo indispensable el diagnóstico diferencial con otras distrofias retinianas.
BIBLIOGRAFÍA
Gass JDM. Heredodystrophic disorders affecting the pigment epithelium and retina. In: Gass JDM.Stereoscopic atlas of macular diseases:diagnosis and treatment on CD ROM. St Louis: Mosby; 1999. Vol. 2, chap 5.
Lopez PF, Aaberg TM. Phenotypic similarities between Stargardt´s flavimaculatus and pattern dystrphies. Aust NZ J Ophthamol 1992; 20: 163-171.
Weleber RG,Carr RE,Murphey. Phenotypic variation including retinitis pigmentosa, pattern dystrophy and fundus flavimaculatus in a single family with a deletion of codon 153 or 154 of the pheripherin/RDS gene. arch. Ophthalmol 1993; 111: 1531-1542.
Benítez del Castillo JM, Trujillo MJ, Del Río T, García B, Ayuso C, García Sánchez J. Retinosis pigmentaria, distrofia patrón y fundus flavimaculatus no relacionadas con una mutación en el gen de la rodopsina,periferina/RDS y ROM 1. Arch Soc. Esp. Oftalmol 2000; 75: 281-285.
Salvatore Daniele, Angelo Carbonara, Claudia Daniele, Gabriella Restagno, Fabrizio Orcidi. Pattern dystrophies of the retinal pigment epithelium. Acta Ophthalmol Scand 1996; 74: 51-55.
Michael F Marmor, J. Arch Mc Namara. Pattern Dystrophy of the retinal pigment epithelium and Geographic Atrophy of the macula.. Am J Ophthalmol 1996; 122: 382-392.
P J Francis, DW Schultz, AM Gregory, MB Schain, R Barra, J Majeswski, J Ott, T Acott, RG Weleber, ML Klein.Genetic and phenotypic heterogeneity in pattern dystrophy. Br J Ophthalmol 2005;89: 1115-1119.