
SEMINARIO DE CASOS CLÍNICOS
DEL-HIERRO-ZARZUELA A1, SANTOS-BUESO E2, DÍAZ-VALLE D2, BENÍTEZ-DEL-CASTILLO JM2
Unidad de Superficie en Inflamación Ocular.
Hospital Clínico San Carlos. Madrid. España.
(1) Licenciado en Medicina. Hospital de Santa Bárbara. Puertollano. Ciudad Real.
(2) Doctor en Medicina.
Publicado parcialmente en Inflamación y alergia ocular (2005).
Presentado parcialmente en el 81 Congreso de la SEOS (Zaragoza 2005).
Presentado en el Seminario de Oftalmología Ramón Castroviejo de la Universidad
Complutense de Madrid (2006).
RESUMEN
Casos clínicos: La amiloidosis es una enfermedad infrecuente que consiste en el depósito a nivel extracelular de la proteina fibrilar amiloide en una o más localizaciones del organismo. Presentamos una serie de tres casos de amiloidosis con distinta afectación ocular.
Discusión: La predilección por las distintas estructuras oculares depende del tipo de sustancia amiloide. Existen formas de amiloidosis primarias de la conjuntiva o de la córnea que generalmente no se relacionan con enfermedad sistémica. Cuando existe afectación orbitaria, palpebral, coroidea o del vítreo es frecuente que el estudio sistémico revele amiloidosis generalizada, generalmente asociada a mieloma múltiple.
Palabras clave: Amiloidosis ocular, amiloidosis conjuntival primaria, distrofia reticular.
INTRODUCCIÓN
Se denomina amiloide al depósito extracelular de una sustancia que al microscopio óptico aparece como eosinófila o hialina y que adquiere la característica tintorial de teñirse con el rojo congo, y de presentar una birrefringencia en verde al exponerse a la luz polarizada. Estas características tintoriales se las confiere el característico patrón de plegamiento beta del componente proteico y mayoritario de la sustancia. Clásicamente las amiloidosis se han clasificado por la afectación sistémica o localizada a que dan lugar, aunque últimamente se tiende más a relacionar con el tipo de proteína que la ocasiona (1) (tabla 1).
A nivel ocular, la amiloidosis puede ser asintomática o manifestarse como síndrome de ojo seco, como alteración de la motilidad, como efecto masa... dependiendo del tipo de estructruras que infiltre. En la amiloidosis primaria generalizada y la asociada al mieloma múltiple, el amiloide puede depositarse en cualquiera de las estructuras, con menor frecuencia en vítreo y vasos retinianos. Es típica la presencia de equimosis y hematomas periorbitarios (2). La amiloidosis secundaria generalizada se observa en infecciones o inflamaciones crónicas, con manifestaciones oculares similares.
En las amiloidosis heredofamiliares, pueden afectarse todas las estructuras oculares, excepto en la tipo III (Iowa). Es muy típica la imagen de pseudopodia lentis que genera el depósito de amiloide detrás del cristalino. En la tipo I (Portuguesa) y II (Indiana) se afecta típicamente el vítreo. En la polineuropatía amiloidótica familiar tipo IV, también denominada síndrome de Meretoja, el cuadro sistémico se combina con una distrofia corneal de patrón reticular; el tipo de proteína es la gelsolina y se ha identificado la mutación responsable en el cromosoma 9q34 (3). La afectación puede ser secundaria a otra patología oftalmológica como triquiasis, queratitis intersticial, trachoma... o primaria.
Entre las formas localizadas destacan la amiloidosis conjuntival (bulbar, límbica o caruncular) y la corneal (distrofia corneal tipo I de Biber-Haab-Dimmer, tipo II, gelatinosa en gotas, de Avellino, subepitelial familiar y degeneración amiloide polimorfa).
En el presente estudio se presentan tres casos clínicos ilustrativos de esta patología.
CASOS CLÍNICOS
Caso Clínico 1
Mujer de 65 años que consultó por ojo izquierdo rojo, con picor y escozor, en cuadros autolimitados periódicos de una semana de evolución en los últimos tres meses. Se encontraba en seguimiento en la unidad de Diagnóstico Precoz de Glaucoma. No presentaba ningún antecedente personal ni familiar de interés. En la exploración la agudeza visual (AV) era de la unidad en ambos ojos y en la lámpara de hendidura se apreciaba una masa subconjuntival rosada, gelatinosa y muy vascularizada (fig. 1). De forma semilunar, se extendía desde la carúncula y la mitad inferior de la conjuntiva bulbar, por todo el fondo de saco inferior de predominio en la mitad interna. El resto de la exploración era normal, incluido el polo anterior y el fondo de ojo. La presión intraocular (PIO) era de 23 mmHg en ambos ojos. El resultado de la biopsia excisional fue informada como depósito de material eosinófilo, acelular y amorfo, rojo congo positivo y birrefringente bajo luz polarizada (figs. 2 y 3). La paciente fue diagnosticada de amiloidosis conjuntival y fue enviada al Servicio de Medicina Interna para valoración sistémica, que resultó negativa, concluyendo el diagnóstico de amiloidosis primaria conjuntival. Cuatro meses más tarde el paciente experimentó un cuadro similar en el ojo adelfo que requirió extirpación.
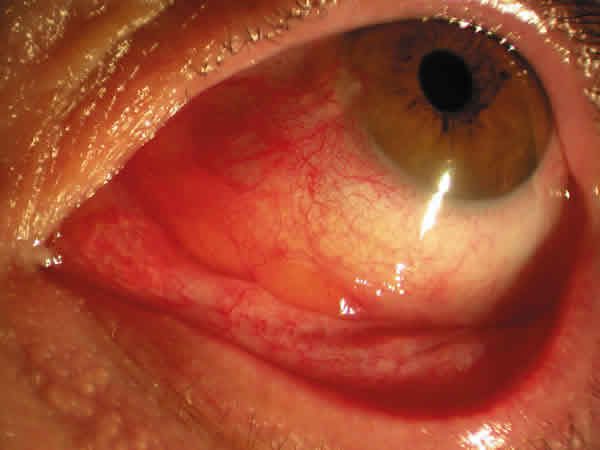
Fig. 1. Caso 1. Masa subconjuntival gelatinosa de
forma semilunar que se extiende desde la carúncula por el fondo de saco
inferior.
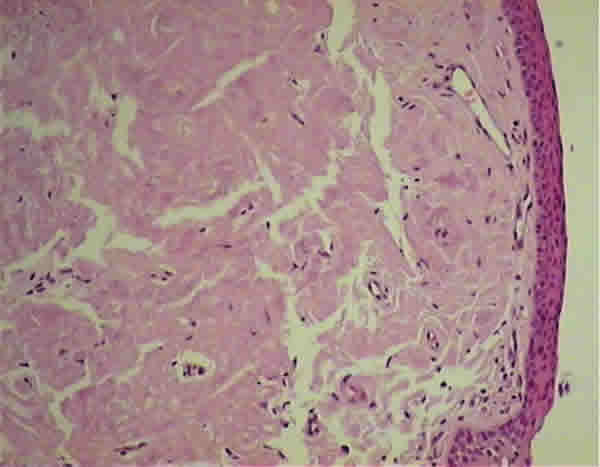
Fig. 2. Caso 1. Tinción hematoxilina-eosina.
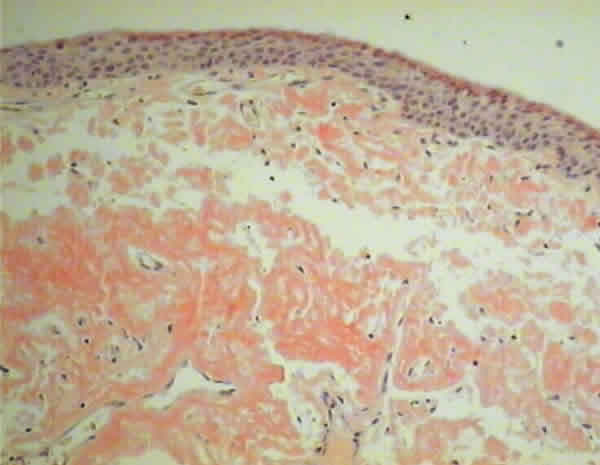
Fig. 3. Caso 1. Tinción con rojo congo.
Caso Clínico 2
Mujer de 62 años sin antecedente personales ni familiares de interés, que consultó por una lesión rojiza en conjuntiva bulbar temporal, de un mes de evolución. La lesión se extendía hacia posterior por el trayecto del músculo recto lateral, (fig. 4) cuya infiltración fue confirmada por resonancia magnética nuclear. La AV era de 0,8 en ambos ojos y el resto de la exploración incluyendo polo anterior, PIO y fondo de ojo fueron normales. Se realizó biopsia de la lesión cuya a anatomía patológica reveló la presencia de amiloide. El estudio sistémico en el Servicio de Medicina Interna resultó negativo. Fue intervenida mediante excisión y cubierta la lesión con membrana amniótica (fig. 5).

Fig. 4. Caso 2. Masa subconjuntival temporal que infiltra el músculo recto
lateral externo.
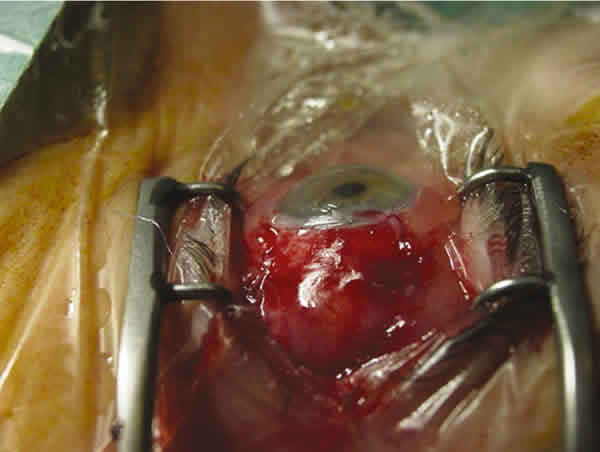
Fig. 5. Caso 2. Masa subconjuntival temporal que infiltra el músculo recto
lateral externo. Aspecto del postoperatorio inmediato con membrana amniótica.
Caso Clínico 3
Varón de 30 años sin antecedentes personales ni familiares de interés, que consultó por erosiones corneales recidivantes. La exploración biomicroscópica reveló una distrofia corneal caracterizada por líneas grisáceas bilaterales, finas en estroma anterior compatibles con una distrofia reticular (fig. 6). El resto de la exploración, incluido el resto del polo anterior, la PIO y el fondo de ojo fueron normales. El estudio sistémico en el Servicio de Medicina Interna resultó negativo.
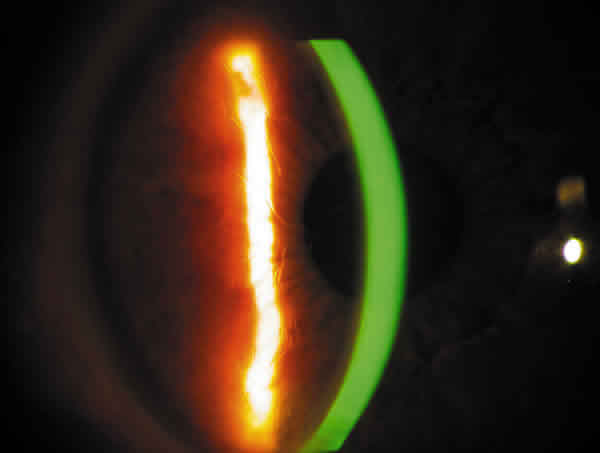
Fig. 6. Distrofia reticular.
DISCUSIÓN
En este artículo presentamos tres pacientes con amiloidois ocular, afectándose diferentes estructuras oculares.
El primer caso corresponde a un cuadro de amiloidosis conjuntival primaria. Se suele manifestar como una masa amarillenta que nace del fórnix y se extiende a conjuntiva bulbar y tarsal. El aspecto macroscópico puede simular otras patologías conjuntivales, como linfomas, papilomas, pseudoterigiones (4) ... La amiloidosis conjuntival suele presentarse en la quinta década de la vida. Típicamente es unilateral, aunque existen casos de bilateralidad, como el caso que presentamos (5).
La afectación de HTO no es exclusiva de formas vítreas sino que en formas localizadas también puede tener lugar. Parece ser debido a la infiltración del ángulo por el amiloide (6).
El segundo caso corresponde a un patrón conjuntival primario con infiltración muscular. Por el momento no existe tratamiento disponible para la amiloidosis, salvo la excisión quirúrgica, con imposibilidad de hacerla completa en ocasiones, como en este caso.
Por último el tercer caso corresponde a un cuadro de amiloidosis corneal. La córnea puede verse afectada en amiloidosis primarias tipo AL, mieloma múltiple, secundarias tipo AA, dentro del cuadro de la polineuropatía amiloidótica familar tipo IV o síndrome de Meretoja, o afectarse exclusivamente la córnea sin estarlo otras estructuras, como Distrofia reticular tipo I o de Biber-Habb-Dimmer, tipo III, distrofia gelatinosa en gotas, distrofia reticulogranular de Avelino o degeneración amiloide polimorfa (7).
En conclusión, el diagnóstico de amiloidosis se confirma siempre mediante anatomía patológica. En todos los casos se debe descartar patología sistémica, a través de analítica, estudio inmunoelectroforético y biopsia de la grasa abdominal.
BIBLIOGRAFÍA
Peter D. Gorevic, Merlyn M. Rodrigues. Ocular amyloidosis. Am J Ophthalmology 1994; 117: 529-531.
Sánchez Salorio M, Díaz Llopis M, Benítez del Castillo JM, Rodríguez Ares MT. Enfermedades endrocrinas y metabólicas. En: Manifestaciones oftalmológicas de las enfermedades generales. LXXVII Ponencia de la Sociedad Española Oftalmología 2001; Cap 9: 328-330
Ola Sandgren. Ocular Amyloidosis, with special reference to the hereditary forms with vitreous involvement. Surv Ophthalmol 1995; 3: 173-190.
Santos-Bueso E, del-Hierro-Zarzuelo A. Amiloidosis primaria conjuntival. Inflamación y alergia ocular 2005; 8: 19-20.
Sáinz Esteban A, Saornil Alvarez MA, Méndez Díaz MC, Blanco Mateos G. Amiloidosis primaria conjuntival: análisis de dos casos. Arch Soc Esp Oftalmol 2005; 80: 49-52.
Gregory A. Nelson, Deepak P. Edward, Jacob T. Wilensky. Ocular Amyloidosis and secondary glaucoma. Ophthalmology 1999; 106: 1363-1366.
Barraquer R. , De Toledo M. , Torres E. Distrofias y degeneraciones estromales. En: Distrofias y degeneraciones corneales. Ed Expas 2004; cap 5: 133-150.