
REVISIÓN ACTUALIZADA
SAMPEDRO LÓPEZ A1, MORÁN M2, PINTO BLÁZQUEZ J3, BARBÓN GARCÍA JJ1, SÁNCHEZ GARCÍA FJ2, ABELAIRAS GÓMEZ V1
Servicio de Oftalmología. Hospital San
Agustín. Avilés (Asturias).
1 Licenciado en Medicina.
2 Licenciado en Medicina. Servicio de Hematología.
3 Doctor en Medicina. Servicio de Anatomía Patológica.
RESUMEN
La clasificación de las neoplasias linfoides siempre ha resultado bastante confusa debido a la gran variedad clínica y a los continuos cambios que ha introducido el desarrollo científico, sobre todo en su caracterización molecular. La actual clasificación de la OMS representa un paso más en el conocimiento de estos tumores al integrar las características anatomopatológicas, inmunológicas y moleculares de las células.
Prácticamente casi todas las proliferaciones linfoides que afectan al globo ocular son linfomas no Hodgkin de células B, siendo las más características el linfoma MALT (por su frecuencia) y el linfoma intraocular primario (el diagnóstico diferencial más importante en las uveítis posteriores), pero también se puede afectar por una hiperplasia linfoide en la conjuntiva y por otras neoplasias linfoides como una infiltración uveal linfoide, linfoma folicular, linfoma de células del manto, plasmocitoma, linfoma B difuso de células grandes y el linfoma T/NK. En esta revisión se repasan los principales rasgos de estos linfomas con una somera descripción de sus características biológicas y clínicas.
INTRODUCCIÓN
Las lesiones linfoides que se localizan específicamente en el globo ocular, y que normalmente se incluyen en el amplio grupo de las orbitarias, presentan una escasa casuística con no muchas variedades clínicas. Estas lesiones linfoproliferativas pueden ser hiperplasias linfoides o verdaderos linfomas, y dentro de ellos bien primarios, como el linfoma MALT (linfoma asociado a mucosas) y el linfoma intraocular primario, o secundarios a una afectación metastásica ocular de un linfoma sistémico, a veces incluso como primera manifestación clínica.
La clasificación vigente REAL/OMS considera las características anatomopatol´ógicas, inmunológicas, citogenéticas y moleculares de las células para establec er grupos pronósticos que ayudan a establecer el riesgo y a orientar el tratamiento. En esta clasificación se reconocen además de los linfomas de Hodgkin, el linfoma no Hodgkin B (tabla 1) y el linfoma T/NK (1). Dentro de los LNH se establece el origen de la neoplasia (en células precursoras y células maduras), en sus distintos estadíos y según su ubicación, en la que el globo ocular formaría parte del sistema extraganglionar (2).
Inmunohistoquímica y biología de los LNH
El diagnóstico histopatológico clásico en las proliferaciones linfoides permitía una clasificación morfológica del tipo celular y generalmente la distinción entre procesos reactivos y tumorales. La inmunohistoquímica supone un gran avance en la tipificación del inmunofenotipo, con un papel fundamental en la clasificación de los linfomas, y la citometría de flujo permite en la mayoría de los casos la determinación del linaje celular, el estadío madurativo y la clonalidad (3). Actualmente las técnicas de citogenética y biología molecular proporcionan además parámetros diagnósticos y pronósticos que condicionan la actitud terapéutica. El grupo de lesiones denominadas hiperplasias linfoides atípicas, que se utilizaba para aquellas proliferaciones intermedias entre hiperplasia linfoide y linfoma pueden concretarse con el estudio genético.
En el estudio genético se pueden utilizar técnicas de citogenética convencional para caracterizar alteraciones cromosómicas estructurales o numéricas y técnicas moleculares: la reacción en cadena de la polimerasa (PCR) y la hibridación in situ (FISH). La PCR permite la ampliación exponencial de un semento de DNA específico y con ello el estudio de la reordenación genética y de las traslocaciones determinando la patología linfoide con una sensibilidad y especificidad cercanas al 100%. Las técnicas de hibridación in situ utilizan sondas de ADN que detectan alteraciones cromosómicas minúsculas. Estos análisis moleculares en los procesos linfoproliferativos permiten establecer el diagnóstico de linfoma, documentan la clonalidad (maligno-clonal vs reactivo-policlonal), la línea celular, demuestran reordenamientos genéticos asociados con subtipos específicos, identifica virus asociados (como el de Ebstein-Barr en el linfoma T/NK) y analiza oncogenes relacionados con progresión y refractariedad al tratamiento como el p53 y el c-myc (1,4).
Linfomas primarios de órbita y globo ocular
El tejido linfoide ocular se encuentra en la órbita, párpados, glándula lacrimal y conjuntiva ocular, a partir del cual se puede originar el linfoma asociado a mucosas (5). Los linfomas extraganglionares primitivos del globo ocular y anexos pueden presentarse con ptosis, proptosis, masas, tumefacción, alteraciones conjuntivales, uveítis o vitritis de larga evolución (4).
El estudio debe incluir anamnesis y exploración sistémica, estudios hematológicos (hemograma, velocidad de sedimentación, estudio de coagulación) y bioquímicos (función renal, hepática, LDH, beta-2 microglobulina, albúmina, preteinograma con inm unoelectroforesis), estudio de médula ósea, de imagen (TAC completo) y LCR si histología agresiva, afectación del SNC, de senos nasales/paranasales, testicular o más zonas extraganglionares. Para su estadiaje se usa la clasificación de Ann Arbor e índice pronósticos entre los que destaca el Internacional Prognostic Index (IPI) (tabla 2).

En muchas ocasiones el diagnóstico histopatológico de estos linfomas es difícil por la calidad de la muestra y se deben realizar técnicas de biología molecular para diagnóstico específico y diferenciar lesiones benignas o pseudotumorales (6-8).
HIPERPLASIAS LINFOIDES
Hiperplasia conjuntival linfoide
Se trata de una proliferación hipercelular benigna rica en linfocitos pequeños y células plasmáticas sin lesiones linfoepiteliales, con cadenas kappa y lambda (policlonales) y Bcl2 negativo, pero morfológicamente indistinguible de un linfoma (9).
La presencia de folículos linfoides con centros germinales no distingue una proliferación benigna de una maligna, y son la inmunohistoquímica y, sobre todo, la biología molecular, los que puede dar el diagnóstico de linfoma (10).
La hiperplasia conjuntival linfoide se suele presentar en adultos, aunque también se describe en niños, sin cambios en la visión o motilidad ocular (10,11). A la exploración se apecia una masa bien delimitada, seudoencapsulada, de color salmón, no adherida a la esclera e indolora (fig. 1). En una reciente revisión sólo un 17% de las lesiones linfoproliferativas conjuntivales son hiperplasias y tanto la localización en fórnix y media periferia conjuntival como la multifocalidad son hallazgos más típidos de linfoma (12). En algún caso de hiperplasia conjuntival linfoide, aunque diagnosticada con criterios histológicos, ha sido documentada una asociación a linfoma sistémico (12).
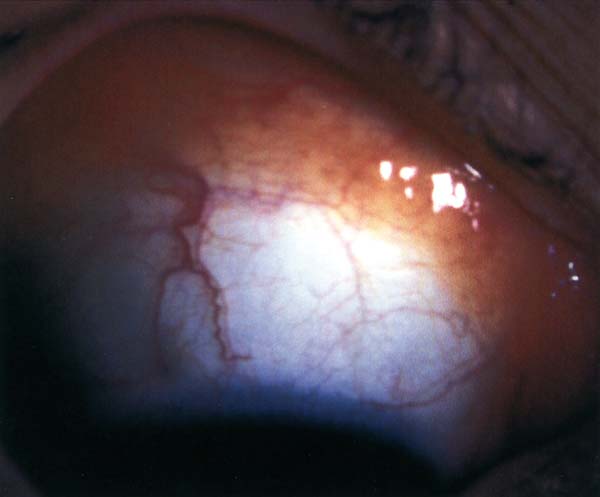
Fig. 1: Hiperplasia conjuntival linfoide. Masa anaranjada en
fondo de saco conjuntival, bien delimitada.
El tratamiento resulta controvertido y hay que considerar la edad del paciente y la extensión de la lesión. Parece indicada inicialmente la biopsia, para estudio por técnicas de PCR, con la exéresis completa de lesiones pequeñas. La observación, sobre todo en ancianos, es una medida lógica pero en pacientes jóvenes se debería considerar la cirugía ante el riesgo futuro de evolución a linfoma, sobre todo ahora que se puede utilizar membrana amniótica para cubrir amplios defectos conjuntivales (13). Los corticoides subconjuntivales perilesionales han sido descritos recientemente como tratamiento (14).
LINFOMAS
Infiltración uveal linfoide
La infiltración uveal linfoide (o hiperplasia linfoide reactiva de la úvea) consiste en una proliferación de lonfocitos y células plasmáticas que se origina primariamente en el tracto uveal, donde sólo existen linfocitos circulantes (4,15).
Clínicamente las lesiones linfoides uveales pueden verse inicialmente como manchas subretinianas amarillentas, y pueden desarrollar a lo largo del tiempo un gran espectro clínico con uveítis, glaucoma, desprendimiento exudativo de retina y proptosis (16,17).
La ecografía y el TAC ayudan al diagnóstico mostrando el engrosamiento coroideo con una baja reflectividad interna, la presencia de nódulos extraesclerales y la ausencia de excavación coroidea. Estos nodulos, si afectan a la conjuntiva bulbar están fijos a la esclera, a diferencia de la movilidad que tienen en los linfomas conjuntivales (18). La biopsia conjuntival resulta significativa si muestra una infiltración estromal de linfocitos con un área subepitelial libre de afectación, lo que sugeriría un origen intraocular (15). La reevaluación de casos antiuos diagnosticados de hiperplasia uveal linfoidea está demostrando que la mayoría eran linfomas de bajo grado (18).
El tratamiento conservador tiene un excelente pronóstico visual utilizando corticoides sistémicos y radioterapia (15). Un pequeño porcentaje de infiltraciones uveales linfoides tienen extensión sistémica, que debe descartarse mediante estudio de imagen torácico y abdominal, estudio hematológico, electroforesis y biopsia de médula ósea (15-17).
Linfomas tipo MALT
Los linfomas del Tejido Linfoide Asociado a Mucosas (MALT) son linfomas B extraganglionares derivados de linfocitos de la zona marginal con alteraciones moleculares que han bloqueado su apoptosis (19). La mayoría de los casos se presenta como una enfermedad localizada, siendo la forma conjuntival menos frecuente que la orbitaria, en este caso se puede hablar de CALT por ser tejido linfoide asociado a la conjuntiva (20). Es preciso descartar que no sea la manifestación de un linfoma sistémico mediante una evaluación completa.
Existe con frecuencia antecedente de enfermedad autoinmune o inflamatoria y se ha relacionado con el virus de la hepatitis C, virus del herpes y clamidias (21).
La lesión más característica es una masa salmón-anaranjada en fondo de saco, no adherida a planos profundos. Si la afectación se extiende a la órbita es frecuente la presencia de ptosis o proptosis (22) (fig. 2). A veces la presencia de puntos blancos coroideos y efusión uveal o un síndrome mascarada intraocular pueden ocular la presencia de un MALT (23,24).
![]()
Fig. 2: Linfoma MALT. Masa asalmonada en conjuntiva bulbar
compuesta por células linfoides que muestran una intensa positividad para el
marcador CD20 (400x).
Las lesiones están compuestas por linfocitos B de la zona marginal, células monocitoides, linfocitos pequeños, células plasmáticas (habitualmente monoclonales) y blastos salpicados. El criterio diagnóstico principal es la presencia de células B monoclonales con expresión de CD20+, CD5, CD10, Bcl16 y ciclina D1 (19).
El tratamiento preconizado va desde la cirugía, quimioterapia sistémica (CHOP o clorambucil más prednisona) o local (con interferón alfa intralesional), a la radioterapia o combinaciones de ellos (25,26). Ha sido también descrita una buena respuesta a antibióticos sistémicos como la doxyciclina (27).
En cuanto al pronóstico la positividad en las células tumorales de antígenos Ki-67 y proteínas p53 guarda correlación con el grado de malignidad (28). Los niveles altos de LDH, el tipo de linfoma y si está localizado o extendido también influyen en la supervivencia (29). La recurrencia de estos tumores suele ser local y el riesgo de presentar afectación extraocular está presente hasta cinco años del diagnóstico (30).
Linfoma intraocular
El linfoma intraocular primario es una variante del linfoma primario del sistema nervioso central, en el que las células linfomatosas invaden retina, vítreo y/o, papila con o sin afectación concomitante del propio SNC (31). El linfoma intraocular tiene una baja supervivencia, tendencia a infiltrar SNC (60-80%) y a ser bilateral (80%) (31,32).
La presentación clínica suele ser una inflamación intraocular con una vitritis tórpida acompañada de una uveítis anterior que responde parcialmente a corticoides y es uno de los clásicos síndromes mascarada (24). En el fondo de ojo pueden aparecer infiltrados subretinianos amarillentos, que en la angiografía pueden mostrar alteraciones del epitelio pigmentario retiniano en forma granular, efecto pantalla e hiperfluorescencia tardía (33) (fig. 3). La ecografía puede detectar condensaciones vítreas, engrosamiento coroideo, aumento del nervio óptico y desprendimiento de retina (34). Si existe afectación del SNC pueden aparecer cefaleas y náuseas por aumento de la presión intracraneal y signos focales como hemiparesia, ataxia y déficit sensitivos (31,35).
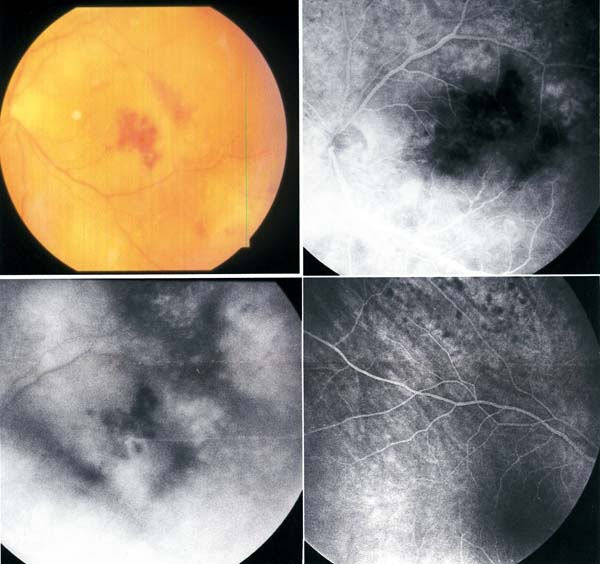
Fig. 3: Linfoma intraocular. Infiltrados subretinianos
amarillentos con efecto pantalla e hiperfluorescencia tardía en la angiografía y
alteraciones granulares del epitelio pigmentario retiniano en el ojo
contralateral.
En cuanto al diagnóstico, ante su sospecha clínica se deben indicar una evaluación neurológica con estudios de neuroimagen (TC o RNM de cráneo y órbita) y una punción lumbar para su examen citológico (36). El hallazgo de células linfomatosas en el líquido cefalorraquídeo (LCR) es diagnóstica de linfoma primario del sistema nervioso central. Si no hay evidencias la siguiente técnica a realizar sería una vitrectomía diagnóstica, aspirando una muestra vítrea sin diluir que debe ser procesada inmediatamente por el patólogo debido a la gran fragilidad de las células linfomatosas (31,37,38). Finalmente se puede recurrir a una biopsia de la lesión, por punción transescleral o transvítrea, o mediante una biopsia coriorretiniana para escindir un bloque de retina-coroides, bien por vía interna o a través de un colgajo escleral (39,40). Si fallan todos los procedimientos diagnósticos se debe considerar la enucleación (31).
El pronóstico es muy pobre y el tratamiento clásico del linfoma intraocular aislado era la radioterapia, aunque actualmente tiende a complementarse con altas dosis de metotrexate ya que atraviesa la barrera hematorretiniana. Las recurrencias oculares aisladas suelen tratarse con metotrexate intravítreo y en caso de asociar extensión al SNC suele asociarse radioterapia del globo con metotrexate sistémico (31).
Linfoma de células del manto
El linfoma de células del manto es un subtipo de linfoma no Hodgkin de fenotipo B, indolente y moderadamente agresivo que afecta a personas mayores. Habitualmente se presenta de forma diseminada con linfadenopatía generalizada, aunque puede asociar o incluso debutar con una afectación extranodal en médula ósea, sangre periférica, bazo, hígado o tracto gastrointestinal (41). En la región periocular y en el propio globo ocular puede aparecer de forma primaria o metastásica (42) (fig. 4).
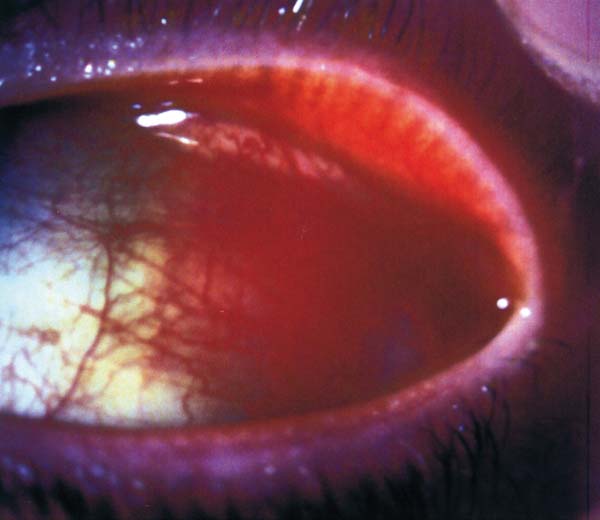
Fig. 4: Masa asalmonada en la conjuntiva bulbar en el contexto
clínico de un linfoma células del manto con afectación sistémica.
Histológicamente puede ser nodular o difuso y hay una variedad típica y otra blastoide, más agresiva. La variedad típica muestra una citología monomorfa, constituida por linfocitos pequeños de núcleo hendido, semejantes a centrocitos. Fenotípicamente se caracteriza por la presencia monoclonal de línea B CD5+, CD10, CD23, frecuentes alteraciones del gen ATM y sobreexpresión de ciclina D1 como consecuencia de la traslocación constante t(11;14) (19). El parámetro pronóstic o con significado adverso más reportado es el índice mitótico elevado (41).
El diagnóstico se basa en el análisis histológico, inmunohistoquímico, y técnicas de biología molecular que permiten identificar la típica traslocación 11:14 (19,43).
La evolución clínica es bastante agresiva con pobre respuesta al tratamiento, siendo la supervivencia media en torno a 3-4 años. El tratamiento consiste normalmente en quimioterapia (prednisona y clorambucil) y radioterapia.
Plasmocitoma y mieloma múltiple
El plasmocitoma es una neoplasia de células plasmáticas (linfocitos B especializados en la producción monoclonal de inmunoglobulinas) que puede aparecer excepcionalmente en la conjuntiva y ser primario (plasmocitoma extramedular solitario) o secundario, asociado a un mieloma múltiple donde la proliferación es sistémica (44). El plasmocitoma solitario no produce una inmunoglobulinemia monoclonal detectable pero requiere vigilancia para descartar la aparición posterior de un mieloma múltiple (4,44). El tratamiento sería la escisión o la radioterapia local (44). La afectación ocular en el contexto de un mieloma requiere terapia sistémica con agentes alquilantes, corticoides y quimioterapia, que se puede completar con radioterapia local (45).
Linfoma folicular
Es el más frecuente de los linfomas indolentes, supone un 30% de los linfomas no-Hodgkin y suele encontrarse diseminado al diagnóstico (46). Histológicamente es un tumor derivado de los linfocitos del centro germinal, con un patrón folicular, que se caracteriza por un inmunofenotipo CD10+ y en casi un 90% de los casos por la sobreexpresión de la proteína Bcl2 secundaria a la traslocación t(14; 18) (4,19). En muchos casos se transforma con el tiempo en un linfoma agresivo de células grandes.
El tratamiento sólo puede ser curativo en estadíos precoces, utilizando radioterapia, mientras que en los estadios tardíos (en los que se suele diagnosticar) ningún tratamiento prolonga de forma significativa la supervivencia, de unos 6-10 años (19). Se utilizan CHOP, CVP, fludarabina y rituximab, un anticuerpo monoclonal aún pendiente de validar resultados de supervivencia a largo plazo.
Linfoma B difuso de células grandes
Este grupo de linfomas, frecuentes en nuestro medio, raramente afectan globo ocular y pueden hacerlo en conjuntiva, iris o como linfoma intraocular primario. Un 30% son secundarios a linfomas preexistentes de bajo grado y un 40% son extranodales, donde puede estar limitado (47).
Las células pueden tener aspectos morfológicos variables, crecen de forma difusa y contienen bastantes mitosis. En la práctica totalidad presentan antígenos B (CD20+) y en un 80% expresión de la proteína Bc16 (19,48).
Los tratamientos quimioterápicos tipo CHOP con o sin radioterapia en campos afectos consiguen respuestas efectivas en un 60-80% de los casos y en muchos la curación.
Linfoma de células T/NK
Los linfomas T/NK constituyen sólo un 10% de los linfomas no Hodgkin y presentan una evolución más agresiva que los linfomas B (1). Normalmente se localizan en la región nasofaríngea (fosas nasales, nasofaringe, orofaringe, cavidad oral, senos paranasales) pero por proximidad hasta un 25% desarrollan una invasión orbitaria o una uveítis/vitritis (antiguo granuloma maligno de la línea media0 (49-51) (fig. 5).
![]()
Fig. 5: Linfoma T/NK. Uveítis anterior con pseudohipopión
recidivante en una paciente con afectación del seno maxilar. Anatomía patológica
con proliferación difusa de células linfoides con disposición angiocéntrica (He,
400x).
En su mayoría derivan de células NK y en menor medida de células T citotóxicas. Se caractriza desde el punto de vista anatomopatológico por presentar angioinvasión y necrosis, y por un inmunofenotipo con positividad CD3 intracitoplasmático, CD2, CD56 (si son células NK) y expresión del virus de Ebstein-Barr (52).
El pronóstico es malo con un curso clínico muy agresivo y una supervivencia corta (53). El tratamiento se basa en una combinación de radioterapia, que a menudo requiere sacrificar el globo ocular, y poliquimioterapia (49).
BIBLIOGRAFÍA
Fraga F, García JF. II Curso de diagnóstico en patología hematolinfoide. Santiago de Compostela 2005. Red de Investigación de Linfomas.
Arranz Sáenz R, Martí Ballesteros E, Osorio Prendes S. Tratamiento de los linfomas no Hodgkin agresivos. In: Fernández-Rañada de la Gándara, Alegre Amor A. Terapia en Oncohematología. 3.ª ed. Madrid: Elsevier SA; 2004; 204-216.
Muñoz Calleja C, Loscertales Puedo J, Gómez Reyno Carnota F. Aplicación clínica de la citología y del inmunofenotipaje en Oncohematología. In: Fernández-Rañada de la Gándara, Alegre Amor A. Terapia en Oncohematología. 3.ª ed. Madrid: Elsevier SA; 2004; 56-65.
Jakobiec FA, Nelson D. Lymphomatous, plasmacytic, histiocytic and hematopoietic tumors of the orbit. In: Duane's Ophthalmology on CD Rom. Philadelphia: Lippincott-Raven Publishers; 2003; vol 2. chap. 39.
Díaz Salas CM, Rodríguez Brito O, Barroso Álvarez MC, Vázquez Olazábal E, Alsina Sarmiento S. Linfomas no Hodgkin de localización orbitaria. Rev Cub Oncol 2000; 16: 79-87.
Coupland SE, Krause L, Delecluse HJ, Anagnostopoulos I, Foss HD, Hummel M. Lymphoproliferative lesions of the ocular adnexa. Analysis of 112 cases. Ophthalmology 1998; 105: 1430-1441.
Avendaño J, Silva M. Linfoma conjuntival. Anales de la Facultad de Medicina. Universidad Nacional Mayor de San Marcos, 2000; vol. 61: 324-326.
Baehring JM, Androudi S, Longtime JJ, Betensky RA, Sklar J, Foster CS, Hochberg FH. Analysis of clonal immunoglobulin heavy chain rearrangements in ocular linfoma. Cancer 2005; 104: 591-597.
Ryan S, Zimmerman LE, kin FM. Reactive linphoid hyperplasia. An unusual form of intraocular pseudotumor. Trans Am Acad Ophthalmol Otolaryngol 1972; 76: 652-671.
Lanuza García A, López-Ramos AL, Pinto Bonilla JC, Rodríguez Pereira C, Cortés Vizcaíno V. Manejo de las hiperplasias linfoides orbitarias. Arch Soc Esp Oftalmol 2005; 80: 353-358.
McLeod SD, Edward DP. Benig lymphoid hiperplasia of the conjunctiva in children. Arch Ophthalmol 1999; 117: 832-835.
Shields CL, Shields JA, Carvalho C, Rundle P, Smith AF. Conjunctival lymphoid tumors. Clinical analysis of 117 cases and relationship to systemic lymphoma. Ophthalmology 2001; 108: 979-984.
Espana EM, Prabhasawat P, Grueterich M, Solomon A, Tseg SCG. Amniotic membrane transplantation for reconstruction after excision of large ocular surface neoplasias. Br J Ophthalmol 2002; 86: 640-645.
Telander DG, Lee TZ, Pambuccian SE, Huang AJW. Subconjuntival corticosteroids for benig lymphoid hiperplasia. Br J Ophthalmol 2005; 89: 770-771.
Grossniklaus HE, Martin DF, Avery R, Shields JA, Shields CL, Kuo IC et al. Uveal lymphoid infiltration. Report of four cases and clinicopathologic review. Ophthalmology 1998; 105: 1265-1273.
Barbón García JJ, Viña Escalar C, Menéndez Fernández CL, Fernández Álvarez C, Carballo Fernández C, Villareal Renedo P. Infiltración uveal linfoide con extensión sistémica. Arch Soc Esp Oftalmol 2003; 78: 173-176.
Ciulla TA, Bains RA, Jakobiec FA, Topping TM, Gragoudas ES. Uveal lymphoid neoplasia: a clinical-pathologic correlation and review of the early form. Surv Ophthalmol 1997; 41: 467-476.
Cockerham GC, Hidayat AA, Bijwaard KE, Sheng ZM. Re-evaluation of «reactive lymphoid hyperplasia of the uvea». An inmunohistochemical and molecular analysis of 10 cases. Ophthalmology 2000; 107: 151-158.
García JF, Piris MA, Morente MM. Procesos linfoproliferativos no Hodgkin de células B. Rev Esp Patol 2004; 37: 139-158.
Zinzani PL, Magagnoli M, Galieni P, Martelli M, Poletti M, Zaja F. Nongastrointestinal low-grade mucosa-associated lymphoid tissue lymphoma: analysis of 75 patients. J Clin Oncol 1999; 17: 1254-1258.
Ferreri AJ, Guidoboni M, Ponzoni M, De Conciliis C, Dell'oro S, Fleischlauer K. Evidence for an association between chlamydia psittaci and ocular adnexal lymphomas. J Natl Cancer Inst 2004; 96: 571-573.
Gili Manzanaro P, Jiménez Mateo-Sidrón V, Gómez Fernández T, Matilla Rodríguez A, Durán Bóveda S, Zato Gómez de Liaño MA. Linfoma primario conjuntival tipo MALT. Studium Ophthalmologicum 1997; 15: 369-371.
Sarraf D, Jain A, Dubovy S, Kreigr A, Fong D, Paschal J. Mucosa-associated lymphoid tissue lymphom with intraocular involvement. Retina 2005; 25: 94-98.
Rothova A, ooijman F, Kerkhoff F, Van der Lelij A, Lokhorst HM. Uveitis masquerade syndromes. Ophthalmology 2001; 108: 336-399.
Baldini L, Blini M, Guffanti A, Fossati V, Colombi M, La Targia ML. Treatment and prognosis in a series of primary extranodal lymphomas of the ocular adnexa. Annals of Oncology 1998; 9: 779-781.
Galieni P, Polito E, Leccisotti A, Martota G, Lasi S, Bigazzi C. Localized orbital lymphoma. Haematologica 1997; 82: 436-439.
Abramson DH, Rollins I, Coleman M. Periocular mucosa-associated lymphoid/low grade lymphomas: treatment with antibiotics. Am J Ophthalmol 2005; 140: 729-730.
Auw-haedrich C, Coupland SE, Kapp A, Schmitt-Gräff A, Buchen R, Witschel H. Long term outcome of ocular adnexal lymphoma sybtyped according to the REAL classification. Br J Ophthalmol 2001; 85: 63-69.
Nakata M, Matsuno Y, Katsumata N, Takenaka T, Kobayashi Y, Narabayashi M. Histology according to the revised European-American lymphoma classification significantly predicts the prognosis of ocular adnexal lymphoma. Leukemia and Lymphoma 1999; 32: 533-543.
Acero Peña A, Domingo Gordo B, Arrevola Velasco L, Gómez García J, Martínez Montero JC. Linfoma de la zona marginal de la conjuntiva: características clínico-patológicas. Arch Soc Esp Oftalmol 2000; 75: 477-480.
Chi-Chao C, Wallace J. Intraocular lymphoma: update on diagnosis and management. Cancer Control 2004; 11: 285-295.
Díaz-Valle D, Migueles Sánchez R, Toledano N. Síndromes mascarada en uveítis. In: Gegúndez Fernández JA. Aproximación clínica al diagnóstico de las uveítis. MacLine, S.L. 2002. Comunicación del 78 Congreso de la Sociedad Española de Oftalmología.
Vélez G, Chi-Chao C, Csaky KG. Fluorescein angiographic findings in primary intraocular lymphoma. Retina 2002; 22: 37-43.
Ursea R, Heinemann MH, Silverman Rh, Silverman RH, DeAngelis LM, Daly SW, Coleman DJ. Ophthalmic, ultrasonographic findings in primary intraocular lymphoma with ocular involvement. Retina 1997; 17: 118-123.
Whitcup SM. Los síndromes mascarada. In: Alió J, Ruiz Moreno JM, Carreras B. Inflamaciones Oculares. Barcelona: Edika Med; 1995; 327-334.
Akpek EK, Ahmed I, Hochberg FH, Soheilian M, Dryja TP, Jakobiec FA, Foster S. Intraocular-Central nervous system lymphoma. Clinical features, diagnosis and outcomes. Ophthalmology 1999; 106: 1805-1810.
Díaz-Valle D, Díaz-Valle T, Toledano Fernández N, García Cruz, Fernández Aceñero MJ. Linfoma intraocular de evolución atípica. Studium Ophthalmologicum 2000; 19: 251-253.
Adan A. Cirugía vitreorretiniana y uveítis. In: Corcóstegui B, Adán A, García-Arumí J, Mateo C, Nieto I. Cirugía vitreorretiniana. Indicaciones y técnicas. Tecnimedia ed; 1995; 299-315.
Cano Parra J, Quijada González A. Exploraciones complementarias en uveítis. In: Gegúndez Fernández JA. Aproximación clínica al diagnóstico de las uveítis. MacLine SL 2002.
Arévalo JF, Freeman WR. Biopsias coriorretinianas. In: Díaz Llopis M. SIDA en Oftalmología. MacLine SL. 1996; 493-501.
Balagué O, Colomo LL, Campo E. Linfoma de células del manto. Rev Esp Patol 2004; 37: 159-172.
Looi A, Gascoyne RD, Chhanabhai M, Connors JM, Rootman J, White VA. Mantle cell lymphoma in the ocular adnexal region. Ophthalmology 2995; 112: 114-119.
Coffee R, Lazarchick J, Chévez-Barrios P, Howard G. Rapid diagnosis of orbital mantle cell lymphoma utilizing fluorescent in situ hybridization technology. Am J Ophthalmol 2005; 140: 554-556.
Prada Sánchez MC, Pérez Moreiras JV, Pérez Encinas JL, Corteza J, Vanegas P. Leiones linfoproliferativas de los anexos oculares. In: Pérez Moreiras JV, Prada Sánchez MC. Patología Orbitaria. Barcelona: Edika Med; 2002; II: 797-811.
Santos-Bueso E, Clavo-González C, Troyano J, Díaz-Valle D, Saiz M, Benítez-del-Castillo JM, García-Sánchez J. Infiltración ocular en paciente con mieloma múltiple. Arch Soc Esp Otalmol 2005; 80: 725-728.
Harris NL, Jaffe NL, Diebold J, Flandrin G, Muller-Hermelink HK et al. Linphoma classification-from controversy to consensys. The REAL and WHO classification of lymphoid neoplams. Ann Oncol 2000; 11(supl 1): 3-10.
Isaacson PG, Norton AJ. Extranodal lymphomas. 2.ª ed New York: Churchill Livingstone, 1995.
Penny RJ, Blaustein JC, Longtine JA, Pinkus GS. Ki-1 positiv e lardge cell lymphomas, a heterogeneous group of neoplasms. Morphologic, immunophenotypic, genotypic and clinical features of 24 cases. Cancer 1991; 68: 362-373.
Hon CH, Kwok AKH, Shek TWH, Chim JCS, Au WY. Vision-threatening complications of Nasal T/NK lymphoma. Am J Ophthalmol 2002; 134: 406-410.
García-Cosío M, Santón A, Méndez MC, Rivas C, Martín C, Bellas C. Nasopharyngeal/nasal type T/NK lymphomas: analysis of 14 cases and review of the literature. Tumori 2003; 89: 278-284.
Junceda Moreno C, Rodríguez González B, Bernal del Castillo T, Corrales Canel B, Junceda Moreno J. Afectación orbitaria por granuloma maligno de la línea media. Studium Ophthalmologicum 2000; 19: 233-236.
Khosravi Shahi PK, Díaz Muñoz de la Espada VM. Linfoma T/NK extraganglionar tipo nasal: caso clínico y revisión de la literature. An Med Interna 2005; 22: 597-600.
Gómez Codina J. Linfomas B y T: Biología, clínica y tratamiento. 2.ª ed. Madrid: Nova Sidonia Oncologia, 2002.