Fig. 1: OCT OI primer día.
SEMINARIO DE CASOS CLÍNICOS
MORAL CAZALLA R1, BELMONTE MARTÍNEZ J2, MARTÍNEZ RUBIO M3, MOYA MOYA MA4
Hospital General Universitario de Alicante.
Servicio de Oftalmología. Alicante. España.
1 Licenciada en Medicina. Facultativo Residente Oftalmología.
Servicio de Oftalmología.
2 Doctor en Medicina. Jefe de Servicio de Oftalmología. Profesor
titular Oftalmología UMH de Elche.
3 Doctora en Medicina. Facultativo Especialista Oftalmología.
Servicio de Oftalmología.
4 Licenciada en Medicina. Facultativo Especialista Oftalmología.
Servicio de Oftalmología.
RESUMEN
Introducción: El síndrome de Vogt-Koyanagi-Harada (VKH) es una patología multisistémica infrecuente que puede afectar al ojo, sistema nervioso, oído y piel, siendo el primero de ellos el órgano cuyo compromiso puede acarrear secuelas más graves. Una detección y tratamiento precoces son fundamentales para evitar complicaciones oftalmológicas potencialmente devastadoras.
Caso clínico: Se presenta el caso de una mujer de 36 años con pérdida brusca de visión en ambos ojos que desarrolló rápidamente desprendimientos serosos retinianos bilaterales y fue finalmente diagnosticada de síndrome de VKH incompleto. Una sospecha clínica precoz y tratamiento intensivo temprano condujeron a la resolución total en un breve espacio de tiempo.
Palabras claves: Vogt-Koyanagi-Harada, uveomeningitis, desprendimiento seroso retiniano, diagnóstico precoz, tomografía coherencia óptica.
INTRODUCCIÓN
El síndrome de Vogt-Koyanagi-Harada (VKH) es un trastorno multisistémico poco frecuente en la población general en el que pueden verse afectados en grado variable el ojo, sistema nervioso, oídos y piel, siendo no obstante el primero de ellos el órgano cuyo compromiso puede acarrear secuelas más graves.
Su etiología es desconocida, aunque se han descrito asociaciones con ciertos antígenos de histocompatibilidad (1-3) que apuntan hacia un posible origen autoinmune (melanocitos), discutiéndose a su vez la posible predisposición genética y una etiología viral (4-6).
Los criterios diagnósticos de la enfermedad, según la Sociedad Americana de Uveítis (7) son:
1. Ausencia de antecedentes de traumatismo ocular o cirugía.
2. Presencia de tres de los siguientes:
a) Iridociclitis bilateral.
b) Uveítis posterior con DR exudativo.
c) Síntomas de afectación del SNC (hipoacusia/acúfenos, meningismo, alteración de pares craneales) o pleocitosis en LCR.
d) Afectación dermatológica: alopecia, vitíligo, poliosis.
Se pueden diferenciar dos formas: el VKH completo (con los criterios diagnósticos mencionados) y el VKH incompleto o forma uveomeníngea (sin afectación de oído o piel).
CASO CLÍNICO
Mujer de raza blanca de 36 años que acude a urgencias por aparición de un escotoma fijo central en el OI en las últimas 24 horas. Refería cefalea frontal intensa de 2 semanas de evolución acompañada de mareo y visión borrosa en los últimos 3 días. Entre sus antecedentes personales destacaba la toma de anticonceptivos orales, ser fumadora de 1 cigarrillo/día y haber sido diagnosticada previamente de crisis migrañosas sin aura.
En el examen inicial únicamente destacaba una agudeza visual sin corrección de 0,3 en el ojo izquierdo (1 en el derecho) y el hallazgo en fondo de ojo de un desprendimiento retiniano seroso central de pequeño tamaño (fig. 1), objetivado mediante tomografía de coherencia óptica (OCT) con 2 puntos de filtración, de localización subfoveal y parafoveal nasal. Los párpados, motilidad ocular extrínseca e intrínseca, y cifras de tensión ocular resultaron normales. Los medios eran transparentes.
![]()
Fig. 1: OCT OI primer día.
Se deriva a la paciente para valoración neurológica urgente, y se programa para estudio angiográfico. Cuatro días después vuelve a consultar con urgencia por pérdida de visión bilateral. En esos momentos la agudeza visual era de visión de bultos en ambos ojos y la imagen funduscópica sugestiva de neurorretinitis bilateral (fig. 2).
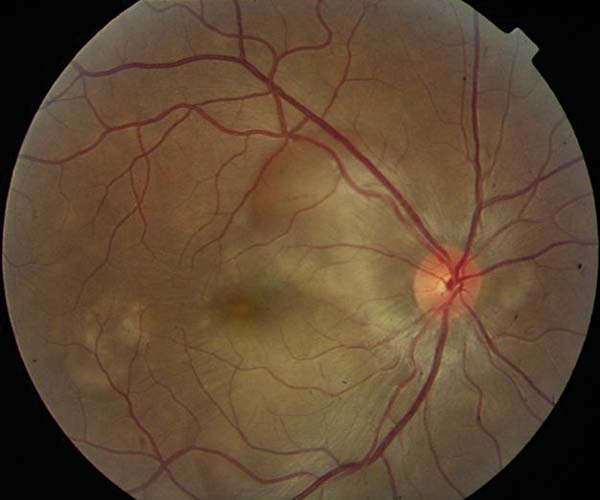
Fig. 2: Fondo de ojo al cuarto día. Aspecto similar en ambos
ojos.
La OCT demostraba la presencia de líquido subretiniano, de nueva aparición en el ojo derecho, y agravado en el izquierdo (fig. 3).
![]()
Fig. 3: OCT OI al cuarto día. Aumento de líquido subretiniano
a nivel macular.
La angiografía fluoresceínica revelaba en ambos ojos varios puntos de filtración en tiempos precoces con importante difusión en los tardíos (figs. 4 y 5).
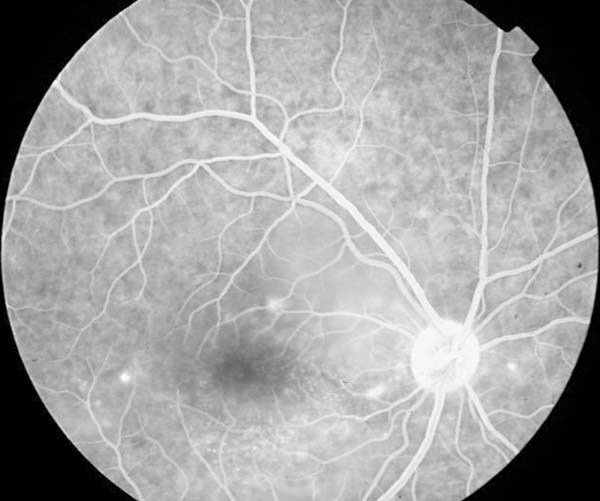
Fig. 4: AGF. Tiempos precoces.
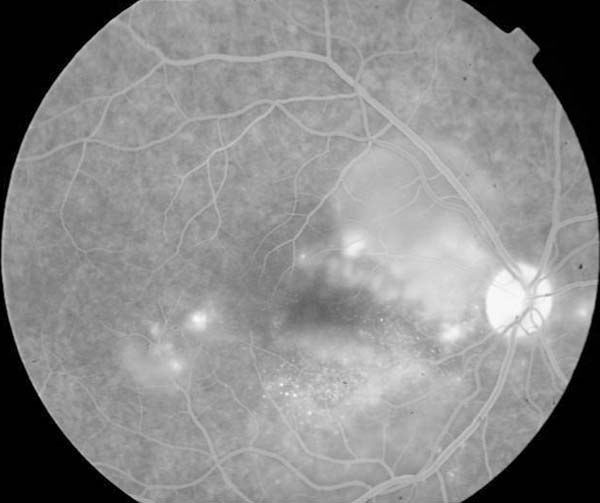
Fig. 5: AGF. Tiempos tardíos.
Ante la sospecha de enfermedad de Vogt-Koyanagi-Harada se decide ingreso hospitalario para realizar punción lumbar, estudio sistémico e inicio de tratamiento corticoideo intensivo, con metilprednisolona i.v. a dosis de 90 mg/día. El TAC craneal fue normal, y el análisis de LCR mostró un aumento de proteínas totales con pleocitosis. No se encontraron otras alteraciones neurológicas. No se evidenció afectación dermatológica, y el examen ORL fue normal. La serología resultó anodina.
A las 48 horas del inicio del tratamiento la visión mejoró espectacularmente, alcanzando una agudeza visual de 1 en el ojo derecho y de 0,8 en el izquierdo, persistiendo en el fondo de ojo tan sólo el desprendimiento seroso a nivel de la arcada temporal inferior del ojo izquierdo. La paciente permanecía apirética, aunque con rigidez y dolor de nuca.
La favorable evolución posterior, con recuperación de la visión (1 en ambos ojos) y resolución de las alteraciones halladas en fondo de ojo y por tomografía de coherencia óptica hicieron posible el descenso gradual de los corticoides hasta llegar a la dosis actual de 20 mg diarios de metilprednisolona oral. Tres meses después del inicio de la sintomatología queda únicamente como secuela una ligera alteración cicatricial (despigmentación) del epitelio pigmentario de la retina que carece de repercusión funcional. La ecografía, aunque fue realizada durante el período de seguimiento, resultó normal.
DISCUSIÓN
El síndrome de VKH es una rara enfermedad caracterizada por una uveítis bilateral con afectación del epitelio pigmentario de la retina y de las meninges, y de forma variable del encéfalo, pares craneales (sobre todo II y VIII), piel y pelo. Con prevalencia e incidencia poco conocidas, muestra especial predilección por los orientales, negros y caucásicos de piel oscura, y es algo más común en mujeres entre la tercera y cuarta décadas. Constituye una causa importante de uveítis autoinmune no infecciosa con desprendimiento de retina secundario, capaz de generar importantes e irreversibles secuelas si no se trata de forma rápida e intensa.
Clásicamente evoluciona en cuatro fases:
Estadio I (prodrómico), con cefalea, febrícula y dolor orbitario.
Estadio II (uveal y neurológico, oftálmico), con uveítis bilateral y visión borrosa súbita, meningismo y alteraciones auditivas, afectación neurológica variable y hallazgos dermatológicos.
Estadio III (convalecencia), con trastornos pigmentarios en el fondo de ojo.
Estadio IV (crónico recurrente o difuso), complicaciones.
Se describen tres tipos de la enfermedad:
I: afectación ocular, sin compromiso de oído o piel.
II: afectación ocular, con al menos una alteración en piel u oídos.
II: afectación ocular, con 2 o más alteraciones en los otros sistemas.
El diagnóstico debe hacerse de forma rápida para comenzar precozmente con un esquema terapéutico intensivo y adecuado, con el fin de intentar evitar una pérdida visual irreversible. No existe hasta el momento una prueba complementaria, a excepción de una angiografía fluoresceínica, capaz de proporcionar una mayor certeza diagnóstica. No obstante no podemos ni debemos olvidar el importante avance que recientemente ha supuesto la aparición de técnicas exploratorias como la tomografía de coherencia óptica, entre cuyas ventajas destacan el ser un método sencillo, rápido e incruento de detección de esta y otras muchas alteraciones del segmento posterior. En el presente caso el empleo de la OCT permitió detectar precozmente la alteración macular, sospechar el diagnóstico e iniciar tratamiento adecuado con corticoides lográndose una rápida remisión. El papel de la ecografía también ha sido destacado, particularmente a la hora de realizar el diagnóstico diferencial con ciertas formas de escleritis posterior (8) aunque en nuestro caso fue normal.
Si bien el período de seguimiento hasta el momento es corto, presentamos un caso de síndrome de VKH incompleto (forma uveomeníngea) como ejemplo de la importancia de la rápida detección (para la cual la OCT resultó fundamental) y del inicio inmediato del tratamiento corticoideo a altas dosis para obtener una buena respuesta al mismo y por lo tanto mayores probabilidades de buen pronóstico funcional.
BIBLIOGRAFÍA
Weisz JM, et al. Association between VKH and HLA DR1 and DR1 in Hispanic patients living in Southern California. Ophthalmology 1995; 102: 1012-1015.
Avellanes García L, et al. HLA DR is strongly associated with VKH disease in Mexican mestizo patients. Ocular Inmunology and Inflammation 1998; 6: 93-100.
Goldberg AC, et al. HLA DRB1 *0405 is the predominant allele in Brazilian patients with VKH. Human Inmunology 1998; 58: 183-188.
Chan C, Whitcup S. M., Nussenblatt R. B. Sympathetic Ophthalmia and Vogt-Koyanagi-Harada Syndrome. Chapter 51.Vol 4. Duane`s Clinical Ophthalmology. 2006. Edit. Tasman W. MD, Jaeger E. A. MD. Ed. Lippincott Williams & Wilkins.
Krashy J, Honzova S. The Vogt-Koyanagi-Harada Syndrome in children. Cesk-Otfalmol 1995; 51: 156-164.
Rutzen A. R., Ortega-Larrolea G., Frambach D.A., Rao N. A. Macular edema in chronic Vogt-Koyanagi-Harada Syndrome. Retina 1995; 15: 475-479.
Read RW, Holland GN, Rao NA, Tabbara KF, Ohno S, et al. Revised Diagnostic Criteria for Vogt-Koyanagi-Harada Disease: Report of an International Committee on Nomenclature. Am J Ophthalmol 2001; 131: 647-652.
Sainz de la Maza MT. ¿Es un síndrome de Vogt-Koyanagi-Harada o una escleritis posterior? Studium Ophthalmologicum 2007; 25: 57-62.