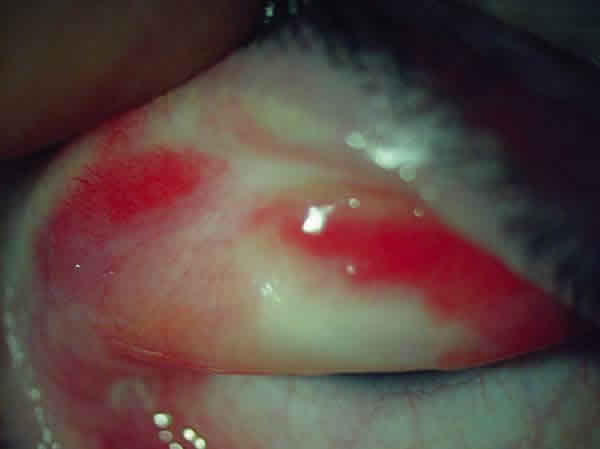
Fig. 1: Sd. Stevens-Johnson, estadio crónico: fibrosis subepitelial conjuntiva tarsal superior.
TOMA DE DECISIONES EN OFTALMOLOGÍA
SAINZ DE LA MAZA MT1
1 Médico Adjunto Consultor. Servicio de Oftalmología. Hospital Clínico de Barcelona. Barcelona.
El eritema multiforme es una erupción dermatológica aguda y autolimitada que, en su forma más leve, el llamado eritema multiforme minor (descrito por Hebra en 1866), afecta a la piel, y en su forma más grave, el llamado eritema multiforme major o síndrome de Stevens-Johnson (descrito por Stevens y Jonson en 1922) afecta a la piel y a las mucosas, incluida la conjuntiva.
Su causa es desconocida pero en la mitad de los casos se puede demostrar un factor precipitante. Dentro de éstos podemos destacar agentes virales, bacterianos, fúngicos, o parasitarios; fármacos orales tales como antibióticos, anti-inflamatorios no esteroideos, anticonvulsivos y fármacos tópicos tales como la tropicamida o la escopolamina; y factores variados tales como las neoplasias, la insolación, el frío, etc. El período de latencia entre la exposición del agente precipitante y el desarrollo de la enfermedad varía desde 1 a 7 días. Se trata de una reacción de hipersensibilidad.
MANIFESTACIONES OFTALMOLÓGICAS
Existe una afectación ocular en el 10% de los pacientes con eritema multiforme minor y en el 90% de los pacientes con síndrome de Stevens-Johnson. La afectación ocular aguda dura de dos a seis semanas. Suelen comenzar como una conjuntivitis purulenta bilateral con edema y/o ulceraciones del párpado. A veces existen ampollas en la conjuntiva y puede haber quemosis. También puede haber una uveitis anterior así como erosiones y úlceras corneales que pueden dar lugar a una perforación corneal. Luego se pasa a un estadio crónico indefinido que en la mayoría de los casos es cicatricial. Puede producirse una fibrosis subepitelial conjuntival (fig. 1) e incluso un simbléfaron (fig. 2) que puede dar lugar a complicaciones posteriores tales como triquiasis, ectropión, entropión, síndrome de ojo seco, lagoftalmos, queratitis por exposición, erosiones y leucomas corneales, queratitis infecciosa, neovascularización corneal, insuficiencia limbar parcial o total, etc. En un porcentaje pequeño de pacientes el estadio crónico indefinido cursa con episodios activos de inflamación que suelen durar entre 1 y 5 semanas; parece ser que pueden ser desencadenados por estrés, menstruación, etc. En estos casos la biopsia conjuntival muestra un depósito de inmunocomplejos en el epitelio y un infiltrado inflamatorio crónico formado por linfocitos T a nivel de la sustancia propia.
Fig. 1: Sd. Stevens-Johnson, estadio crónico: fibrosis subepitelial conjuntiva
tarsal superior.
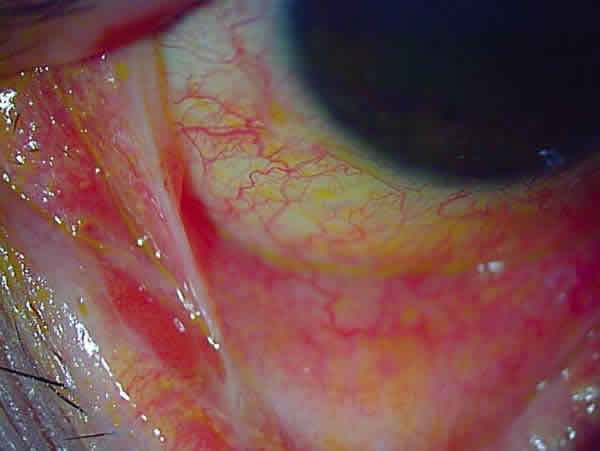
Fig. 2: Sd. Stevens-Johnson, estadio crónico: simbléfaron.
TRATAMIENTO ESTADIO AGUDO (tabla 1)
La utilización de los esteroides sistémicos se basa en el concepto de que el síndrome de Stevens-Johnson está mediado por una reacción de hipersensibilidad tipo IV. Patterson y cols publicaron que los esteroides sistémicos a altas dosis, durante poco tiempo, comenzando entre el día 0 y el día 22 del inicio de los síntomas, habían sido efectivos en el control del estadio agudo.

La utilización de inmunoglobulinas intravenosas puede ser efectiva en controlar la progresión de la enfermedad y en disminuir el tiempo de curación de la piel.
La ciclosporina sistémica también ha demostrado efectividad al inhibir la activación de los linfocitos CD8. Sin embargo, es importante recordar que no comienza a ser terapéuticamente activa hasta el mes del inicio.
La plasmaféresis puede ser beneficiosa al retirar de la circulación sanguínea el fármaco, el metabolito del fármaco, los mediadores citotóxicos, o los anticuerpos. Debido a ello, las secuelas oculares pueden disminuir.
En cuanto a las medidas locales, el recubrimiento de toda la superficie ocular (incluyendo conjuntiva bulbar y tarsal) con membrana amniótica ha demostrado ser efectivo en disminuir la afectación ocular. Debe ser colocada entre los 7 y 10 primeros días del inicio de los síntomas y debe suturarse a los bordes palpebrales con puntos de anclaje en fórnices superiores e inferiores (figs. 3 y 4). La utilización de Prokera (Biotissue, Miami, FL) permite la aplicación de la membrana amniótica sin aplicación de suturas y por tanto sin necesidad de entrar en el quirófano (en la misma cama del paciente) porque se trata de una membrana amniótica que cubre un anillo de simbléfaron (fig. 5). Sin embargo, los resultados no son tan beneficiosos al no conseguir un completo recubrimiento de toda la conjuntiva bulbar y tarsal.
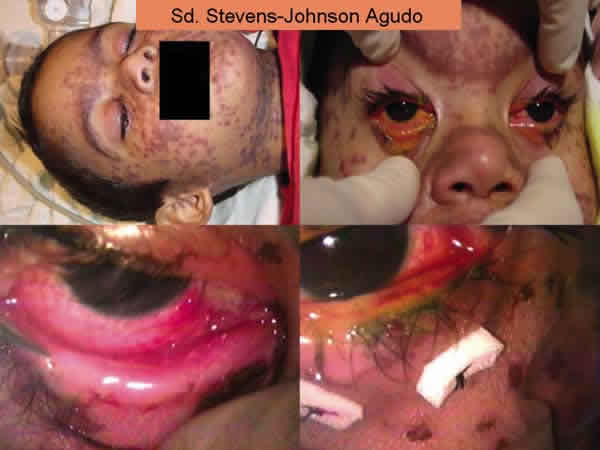
Fig. 3: Sd. Stevens-Johnson, estadio agudo, recubrimiento membrana amniótica
toda la superficie ocular incluyendo la conjuntival bulbar y tarsal.
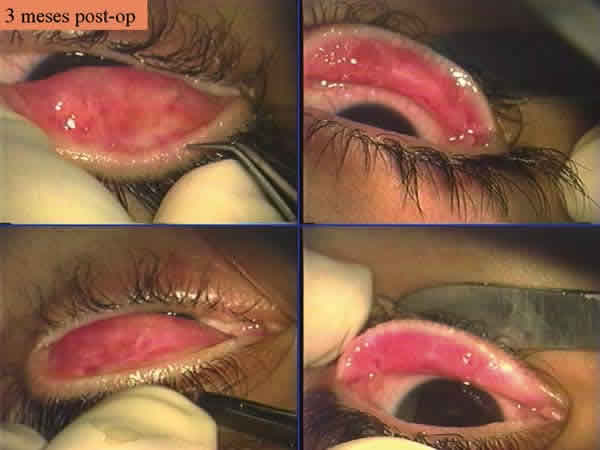
Fig. 4: Tres meses post-op. Existe cierta fibrosis subepitelial tarsal pero no
erosiona la córnea ni produce retracción en los párpados.
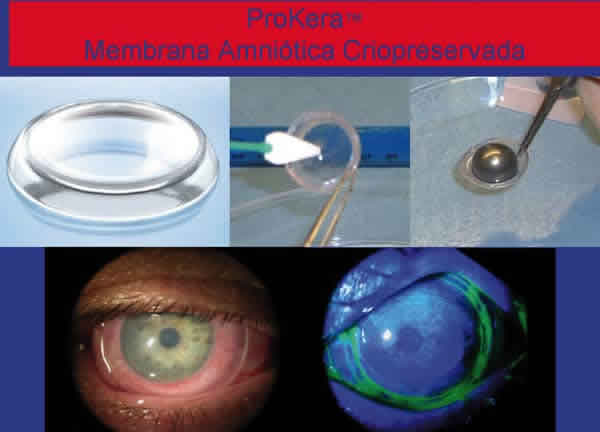
Fig. 5: Prokera (Biotissue, Miami): se trata de una membrana amniótica
criopreservada que se adosa a un anillo de simbléfaron que se coloca en la
superficie ocular y permite la actuación de la membrana amniótica sin necesidad
de suturas.
TRATAMIENTO ESTADIO CRÓNICO (tabla 2)
Debido a la destrucción parcial o total de las células limbares y a la sequedad importante de la superficie ocular, el tradicional transplante de córnea está condenado al fracaso en la mayoría de casos.

El transplante de membrana amniótica para tratar la conjuntivalización corneal o los defectos epiteliales persistentes puede ser efectiva si existen ciertas células limbares funcionantes (insuficiencia limbar parcial). El transplante de membrana amniótica también puede ser efectivo para la reconstrucción del fórnix conjuntival aunque debe realizarse con mitomicina C.
Si existe una insuficiencia límbica total, el alotransplante de células limbares es una posibilidad terapéutica pero los resultados a largo plazo en el síndrome de Stevens-Jonhson son malos a pesar de la inmunosupresión sistémica continua.
El transplante autólogo de células epiteliales orales cultivadas tiene la ventaja de ser tejido autólogo y por tanto no es necesario la inmunosupresión sistémica (fig. 6). En la actualidad se están evaluando los resultados a largo plazo de la supervivencia de las láminas epiteliales mucosas.
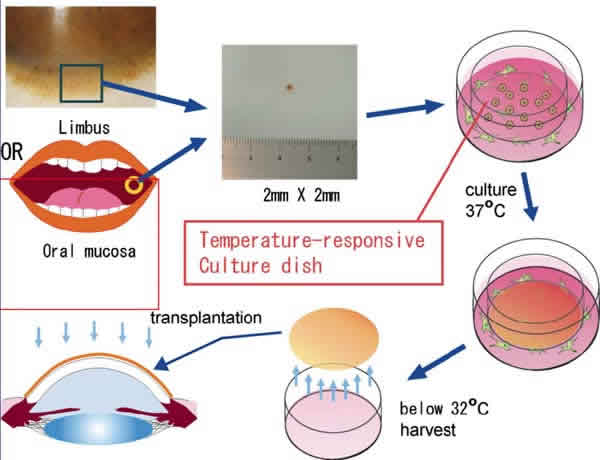
Fig. 6: Transplante autólogo de células orales epiteliales cultivadas.
El transplante de la mucosa del paladar puede ser efectiva en la reconstrucción de los párpados en casos de entropion cicatricial y triquiasis porque aporta soporte estructural además de una superficie mucosa con mínima o nula contracción.
Las lentes de contacto esclerales gas permeables (lente escleral de Boston) reducen el microtrauma asociado a la triquiasis y a los párpados queratinizados, mejoran la visión en las córneas con astigmatismos irregulares y pueden ser utilizadas por ojos con deficiencia lagrimal (a diferencia de las lentes de contacto regulares).
La queratoprótesis constituye la última opción en los casos con enfermedad de la superficie ocular en estadio muy avanzado. La queratoprótesis de Boston es una posibilidad que recientemente ha mejorado su pronóstico a largo plazo desde la introducción de la profilaxis antibiótica continua con vancomicina. La osteo-odonto-queratoprótesis es una técnica quirúrgica muy compleja que también intenta conseguir cierta rehabilitación visual en este tipo de pacientes.
EL FUTURO
Las estrategias terapéuticas, principalmente sistémicas, deben estar dirigidas a detener la progresión de la enfermedad en el estadio agudo y por tanto evitar las secuelas, especialmente oculares. Se requieren estudios multicéntricos, prospectivos, randomizados para la evaluación de ciertos tratamientos, tales como la inmunoglobulina intravenosa o la plasmaféresis.