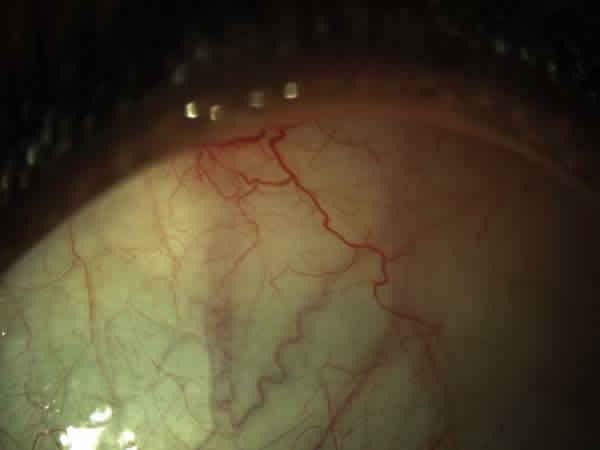
Fig. 1: Tortuosidad de los vasos conjuntivales.
SEMINARIO DE CASOS CLÍNICOS
PÉREZ-ÁLVAREZ MJ1, DÍEZ BIENVENIDO N1, GARCÍA-SÁNCHEZ J
2Hospital Clínico San Carlos. Unidad de
Retina. Madrid.
1
Licenciado en Medicina.
2 Doctor en
Medicina.
RESUMEN
La drepanocitosis es una hemoglobinopatía que cursa con una alteración morfológica de los glóbulos rojos, que adquieren un aspecto semilunar. Estas células anormales sufren mayor hemolisis, y además pueden obstruir los pequeños vasos sanguíneos reduciendo el flujo sanguíneo en los diferentes órganos y en el ojo. Se presenta el caso de una mujer de 19 años, de origen afroamericano, diagnosticada de anemia drepanocítica que presentaba retinopatía proliferativa y que fue tratada con panfotocoagulación bilateral.
Palabras clave: Anemia drepanocítica, neovasos en abanico.
INTRODUCCIÓN
La enfermedad de células falciformes (ECF) es una hemoglobinopatía que se caracteriza por la existencia de eritrocitos en forma de hoz. Dicha deformidad se ve favorecida en condiciones tisulares de baja oxigenación y pH ácido. Los hematíes son más frágiles y proclives a la hemolisis, y además pueden causar obstrucciones de vasos de pequeño calibre. Son frecuentes los fenómenos vasoclusivos en diferentes órganos como bazo, pulmones, riñón y cerebro. También se afectan tejidos con pequeñas arteriolas terminales, entre ellos, la cabeza del fémur y los ojos.
CASO CLÍNICO
Se trata de una mujer de 19 años diagnosticada de anemia drepanocítica. Fue ingresada en sucesivas ocasiones por sepsis por salmonella, neumonía de lóbulo inferior izquierdo. Presentó varios episodios de crisis hemolíticas, durante los cuales la exploración oftalmológica fue anodina. El tratamiento habitual consistió en ciclos de ácido fólico, al que se añadió Hidroxiurea a dosis 0,5 a 1 gr al día, con la intención de reducir las crisis dolorosas.
Durante el último ingreso de hacía 4 meses, debido a una crisis dolorosa en pierna izquierda, en la revisión oftalmoscópica se evidenció una retinopatía proliferativa en ambos ojos.
En la exploración oftalmológica la agudeza visual mejor corregida (AVMC) era de 0,6 en ambos ojos (AO). Por biomicroscopía anterior se apreciaba muy leve tortuosidad de los vasos conjuntivales, sin atrofia iridiana (fig. 1). La tonometría era de 16 mmHg. AO La funduscopía de ojo derecho (OD) mostraba oclusiones vasculares en la periferia 360 grados, con terminación muy abrupta de los mismos, telangiectasias, anastomosis arteriovenosas, zonas de no perfusión capilar, vasos hialinizados (fig. 2). El ojo izquierdo (OI) presentaba además de las lesiones descritas en el OD, neovasos muy llamativo en «abanico» en la zona temporal superior y blanco sin presión en la periferia nasal (fig. 3).
Fig. 1: Tortuosidad de los vasos conjuntivales.
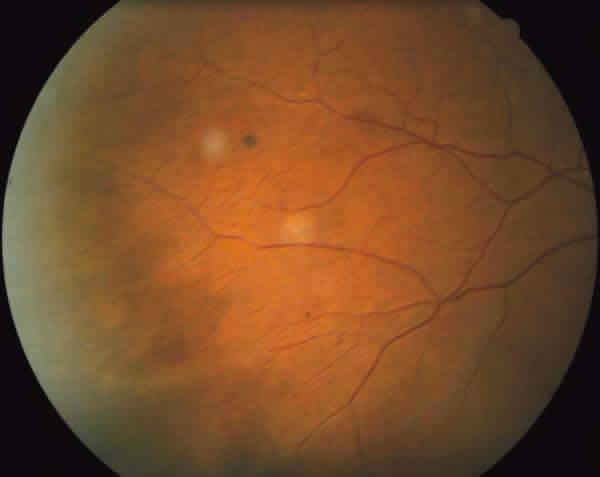
Fig. 2: Funduscopía ojo derecho donde se aprecian áreas de oclusión capilar y
shunt arteriovenosos.
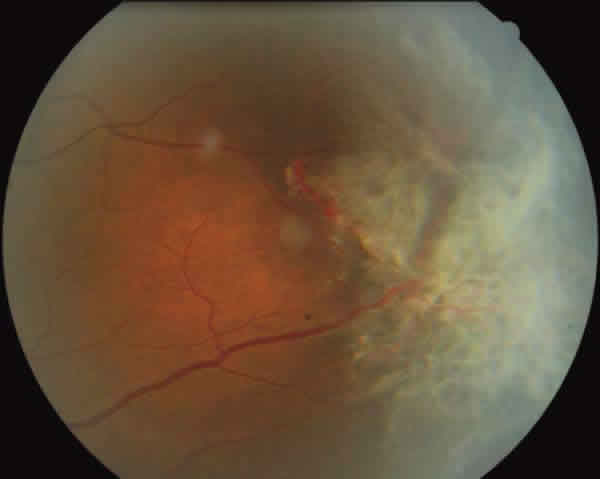
Fig. 3: Funduscopía ojo izquierdo con neovascularización "en abanico".
La angiofluoresceingrafía (fig. 4) evidenció una gran isquemia periférica y los neovasos del OI (fig. 5) pero sin isquemia macular. La tomografía de coherencia óptica tampoco demostró alteraciones foveales.
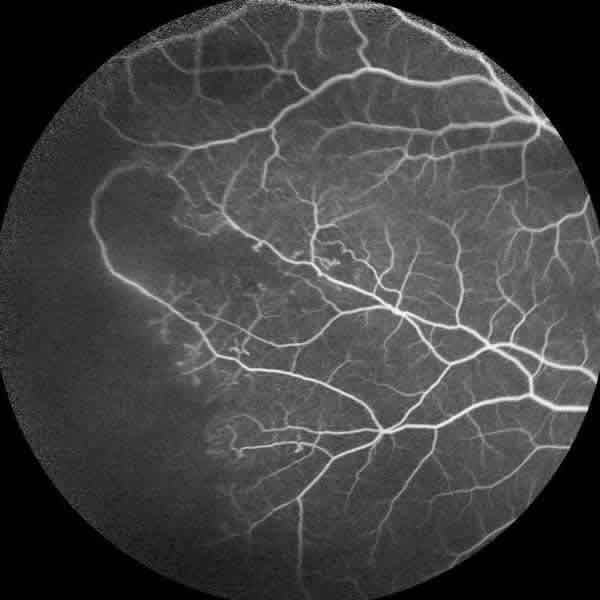
Fig. 4: Angiofluoresceingrafía con extensas áreas de no perfusión capilar
periférica en ojo derecho.
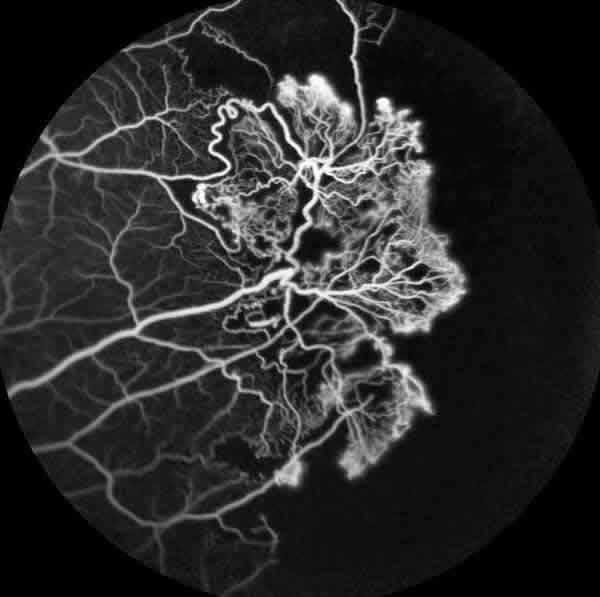
Fig. 5: Angiofluoresceingrafía de ojo izquierdo que muestra el importante
complejo neovascular.
Se realizó una panfotocoagulación bilateral, más intensa en la periferia, y tras un periodo de seguimiento de 2 meses, los neovasos de OI habían regresado sin nuevos signos de neovascularización, manteniéndose la misma AVMC que en el momento del diagnóstico.
DISCUSIÓN
La ECF cursa clínicamente con una anemia hemolítica, congénita y crónica que afecta mayoritariamente a enfermos de raza negra. Su expresividad es variable, desde sujetos con trastornos leves a graves alteraciones. La hemoglobina presenta una mutación en la cadena de globina por la sustitución del péptido en la posición 6, Ácido Glutámico por Valina (Hemoglobina S) o Lisina (Hemoglobina C), en una o ambas cadenas beta. Esta anomalía favorece la polimerización de la molécula cuando se desoxigena (desoxihemoglobina) lo que produce la deformación del hematíe.
En el polo anterior pueden encontrarse modificaciones del aspecto de los capilares conjuntivales en forma de sacacorchos y atrofia iridiana secundaria a oclusión de los capilares del mismo. En el polo posterior podemos encontrar agregados de glóbulos rojos en los pequeños vasos de la superficie de la papila, o un aspecto brillante de la región perimacular por la afectación de las arcadas perifoveales. Las anomalías más evidentes suelen hallarse en la retina periférica, en el área donde el calibre de las arteriolas se hace más pequeño en su alejamiento de la papila. Podemos describir diversos estadios: 1) Oclusiones arteriolares periféricas, 2) Anastomosis arteriovenosas periféricas, 3) Neovascularización prerretiniana, 4) Hemorragia vítrea, 5) Desprendimiento de retina. Por tanto el examen de la retina periférica puede poner en evidencia cualquier aspecto del cortejo semiológico propio de la isquemia retiniana, así pueden percibirse hemorragias rojo-naranja (hemorragias en parche salmón), pequeñas lesiones refringentes (manchas iridiscentes), o manchas pigmentarias periféricas (black sunburst) (1). La incidencia de retinopatía proliferativa en la ECF varía desde 5 a 10%, según el fenotipo, siendo más frecuente en SC que en SS y S-talasemia., por tanto existe una discrepancia entre la severidad de las lesiones sistémicas y las oculares que todavía no podido ser correctamente explicada (1).
Se ha comprobado que el riesgo de retinopatía proliferativa es mayor a partir de los 10 años de edad, y además se va incrementando con la edad (2). Por tanto se recomienda la realización de revisiones bianuales de los 10 a los 20 años de edad. A partir de esta edad deben ser de carácter anual por el mayor riesgo de hemorragia vítrea o desprendimiento de retina. Este hecho se constata en nuestra paciente, que no presentó retinopatía hasta la última revisión periódica a la edad de 19 años.
La tomografía de coherencia óptica mostraba la atrofia que se produce en las capas internas de la retina, con respeto de los fotorreceptores y del epitelio pigmentario. Esto se debe a la oclusión de las arteriolas retinianas. La isquemia macular produce un signo muy característico, denominado depresión macular y aparece como una zona oscura adyacente a la fóvea, no siempre asociada a disminución de AV. En la AFG se objetiva aumento de la zona avascular foveal (4). Nuestra paciente presenta respeto macular, y por tanto mantenía buena AV, a pesar de la intensa isquemia periférica.
El único tratamiento profiláctico de las complicaciones de la enfermedad es la hidroxiurea sistémica (1,2). Sin embargo, una vez establecida la retinopatía proliferativa, la panfotocoagulación retiniana ha demostrado su eficacia en reducción de la incidencia de hemorragia vítrea y desprendimiento de retina (1,3). Por ello, insistimos en la necesidad de revisiones periódicas de estos pacientes debido a la potencial gravedad de las manifestaciones oculares,
BIBLIOGRAFÍA
Emerson GG, Harlan JB, Fekra JS, Lutty GA, Goldberg MF. Hemoglobinopathies. In: Stephen J. Ryan. Retina. St. Louis: Mosby; 200; II: 1283-1296.
Babalola OE, Wambebe CO. When should children and young adults with sickle cell disease be referred for eye assessment? Afr J Med Med Sci. 2001; 30: 261-3.
Seiberth V.Transscleral and transpupillary laser coagulation in proliferative sickle-cell retinopathy. Ophthalmologe. 2001; 98: 199-202.
Witkin AJ, Rogers AH, Ko TH, Fujimoto JG, Schuman JS, Duker JS. Optical Coherence Tomography Demonstration of Macular Infarction in Sickle Cell Retinopathy. Arch Ophthalmol 2006; 124: 746-747.