HISTORIA Y HUMANIDADES
MURUBE E1, ESTEBAN DE ANTONIO M2, MURUBE J
31
Director médico de LasikCenter. Madrid.En la parte I de esta breve historia sobre la genética y la motilidad ocular secuenciamos la exposición de la aparición de los conocimientos de los estrabismos esenciales, las oftalmoplejías familiares aisladas, y los nystagmos. Dedicaremos esta parte II a los defectos oculomotores sindrómicos.
A lo largo de la historia del conocimiento del estrabismo se fueron encontrando múltiples asociaciones sindrómicas, que a menudo posteriormente se fueron disgregando en nuevos síndromes, fenómeno éste que será cada vez más frecuente conforme aumenta el conocimiento de la causa original y su cascada de efectos.
Los trastornos sindrómicos oculomotores podríamos exponerlos ordenadamente según los genes mutados y sus proteínas implicadas, o según su tipo de herencia (autosómica, gonosómica, multifactorial, mitocondrial, recesiva, dominante, etc.), o según los tejidos alterados (nerviosos, musculares, óseos), o la localización del defecto asociado (supranucleares, nucleares, nerviosos periférico, musculares, dismórficos, etc.). Las combinaciones serían tan variadas, y los aspectos aún por descubrir son tantos, que haríamos más confusa la exposición que si lo hacemos por el orden alfabético de los nombres actualmente aplicados a los síndromes.
AICARDI (SÍNDROME DE)
El síndrome de Aicardi se inició en su conocimiento en 1969 (1) y pasó a recibir tal nombre con otra publicación del mismo autor de 1988 (2). Se manifiesta en niñas ya desde los primeros días o semanas de vida. Tiene alteraciones neurológicas como convulsiones y retardo mental, derivadas de lesiones cerebrales entre las que destaca la frecuente agenesia del corpus callosum. El retardo psico-motor en las formas graves apenas permite aprender a andar y hablar. A veces hay anomalías costales y vertebrales. En fundus oculi abundan las áreas redondeadas de atrofia corio-retiniana cercanas a la papila óptica. Los trastornos oculomotores pueden existir, aunque no son frecuentes (112).
El gen causal está localizado en Xp22. Se transmite según el modo dominante ligado al cromosoma X. El síndrome sólo suele observase en niñas, pues es letal para el varón.
ALEXANDER (ENFERMEDAD O SÍNDROME DE)
Es una leucodistrofia que afecta a la envuelta aislante mielínica de las fibras nerviosas del cerebro, especialmente de las astrocíticas descrita hace ya 60 años (4). Se han definido 3 formas: En su forma infantil se asocia a hidrocefalia, retraso mental y espasticidad muscular y manifestaciones oculomotores, y lleva a la muerte en unos meses. Las formas juvenil y adulta suelen tener manifestaciones oculares más notorias, como nystagmus en resorte y nystagmus parético, blefaroptosis, y parálisis facial.
Se debe a una mutación del gen que regula la proteína fibrilar glial ácida (PFGA) astrocitaria. Este gen se localiza en el cromosoma 17 en posición 17q21. Su transmisión es autosómica, recesiva o dominante.
APERT (SÍNDROME DE)
El síndrome de acrocefalo-sindactilia de Apert (8) se describió ya en 1906. Reúne malformaciones esqueléticas como oxicefalia (cráneo en torre), malformaciones digitales simétricas y agenesia de cuerpos vertebrales. A veces hay estrabismo, nystagmus, blefaroptosis superior, exoftalmos e hipertelorismo. Ocasionalmente puede sobrevenir una atrofia del nervio óptico, secundaria a la estrechez del conducto óptico. Suele presentarse desde el nacimiento. Hay cuadros clínicos de agenesias de músculos oculomotores que podrían considerarse formas frustras de síndrome de Apert (9), aunque cuadros similares pueden darse limitados a las agenesia muscular (129,153).
El síndrome de Cole-Carpenter (41) es otra osteogénesis imperfecta, aparentemente similar al síndrome de Apert, con algunas diferencias como hidrocefalia y prognatismo. La analítica genética molecular determinará si tienen el mismo trastorno genético.
El síndrome de Apert se transmite hereditariamente de forma autosómica recesiva, y muy raramente, dominante.
APRAXIA OCULOMOTORA CON ATAXIA
Descrita por Aicardi (2) en 1988, se manifiesta neurológica y oftalmológicamente parecido al síndrome de ataxia-telangiectasia, pero sin déficit inmunitario ni telangiectasias, y algunos clínicos prefieren considerarlo distinto al ya citado síndrome de Aicardi. La apraxia oculomotora se acompaña con movimientos cefálicos parecidos a los de la apraxia de Cogan.
Se transmite por el modo autosómico recesivo. Se debe a una anomalía del gen de la aprataxina (APTX) localizado en el cromosoma 9, en 9p13.3, que activa la hidroxilación y reparación de los filamentoas de DNA.
APRAXIA OCULOMOTORA CONGÉNITA
Vide: Cogan (Síndrome de).
ATAXIA CEREBELOSA HEREDITARIA DE FRIEDREICH
La ataxia de Friedreich (70) (1863) fue la primera ataxia cerebelosa descrita. Es una lesión progresiva de los sistemas nervioso central y muscular, que empieza en la infancia o pubertad, manifestándose por dismetría, desequilibrio, adiadococinesia, disartria, nystagmus, y más raramente por trastornos piramidales y cifoscoliosis dorsolumbar. Acaba obligando a desplazarse en silla de ruedas, y lleva a la muerte en la edad adulta media. Se asocia a veces a oftalmoplejias externas congénitas, nystagmus y distrofia de conos y bastones (102,117).
Afecta a 1 x 50.000 personas, igual a hombres y mujeres. Es de herencia recesiva autosómica. El gen tarado está en el cromosoma 9. Las manifestaciones de la enfermedad se deben a la incapacidad de producir la proteína frataxina.
ATAXIA ESPINO-CEREBELOSA DE MARIE
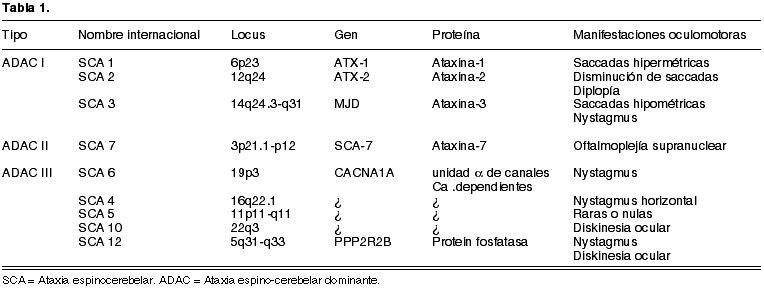
La ataxia de Marie (o de Pierre Marie) (119) es una ataxia espino-cerebelosa, debida a una atrofia progresiva de las capas corticales del cerebelo y de los núcleos dentado y olivar. Se manifiesta porque desde el nacimiento (60), infancia (66) o edades medias de la vida aparece una ataxia espástica de la marcha, dismetría de movimientos e hipertonía. A veces se asocia una oftalmoplejia externa. Arnalich, Rebolleda y Muñoz Negrete han publicado recientemente un caso SCA 7 asociado a distrofia de conos y bastones (10).
En el momento actual son varios los genes tarados encontrados en cuadro clínicos parecidos que podrían ser incluidos en la heredoataxia espino-cerebelosa de Marie, por lo que en breve se espera que se ratifiquen como síndromes distintos (tabla 1).
ATAXIA-TELANGIECTASIA (SÍNDROME DE)
Vide: Louis-Bar (Síndrome de).
BARDET-BIEDL(SÍNDROME DE)
Vide: Laurence-Moon-Bardet-Biedl (Síndrome de).
BASSEN-KORNZWEIG (SÍNDROME DE)
Es una a-b-lipoproteinemia, con incapacidad para absorber y transportar lípidos, con esteatorrea e hipocolesterolemia y con debilidad muscular (18,58). Se asocia raramente a nystagmus, blefaroptosis y oftalmoplejia progresiva.
Se hereda de forma autosómica recesiva.
BLOCH-SULZBERGER (SÍNDROME DE)
También llamado síndrome de Incontinentia pigmenti, es una atrofia cutánea con pigmentación lineal , que se manifiesta al nacer o pocos meses después, y se acompaña de dermatitis, alopecia parcial, anomalías dentales y retraso mental (21,166). En el 25% de los casos hay síntomas oculares como estrabismo, nystagmus, escleras azules, catarata y pseudogliomas retinianos.
El síndrome afecta en el 95% de los casos a hembras, y su transmisión genética es dudosa. El gen responsable de la enfermedad parece estar situado en el brazo corto del cromosoma X, en p11. Cuando se ha determinado una transmisión hereditaria, es ligada al sexo, de modo dominante o recesiva (163) con letalidad para el varón.
Una variedad de incontinentia pigmenti es el síndrome de Naegeli (Vide) (132).
BONNET-DECHAUME-BLANC (SÍNDROME DE)
Este síndrome (24), quizás superponible al más tarde descrito por Wyburn-Mason (190), es una angiomatosis cutánea y retinocerebral, que a veces se manifiesta en los ojos, y cuando lo hace es en forma de estrabismo, nystagmus, blefaroptosis, anisocoria y pérdida de visión.
Se han descrito casos de trasmisión autonómica dominante.
BONNEVIE-ULLRICH (SÍNDROME DE)
Descrito en 1930 por Ullrich (175) y en 1936 por Bonnevie (25), es un trastorno congénito, que se caracteriza por malformación de extremidades (sindactilia, malformaciones ungueales), atrofia muscular, laxitud cutánea, pliegues cutáneos del cuello (pterygium colli), linfoedema de extremidades, y a veces enanismo. Se han citado casos con afectaciones oculares como blefaroptosis, hipoplasia de glándulas lacrimales y ausencia de carúncula y alteraciones de la motilidad ocular.
El síndrome es a menudo hereditario. Así, se han descrito varios casos familiares asociados a paresia bilateral congénita del recto lateral (42).
BROWN (SÍNDROME DE)
Es una fibrosis de la vaina del músculo oblicuo superior, generalmente unilateral. La inextensibilidad del oblicuo superior provoca una impotencia en la contracción del oblicuo inferior homolateral cuando el ojo está en adducción. La hipotropia del ojo afecto, un síndrome en V y el tortícolis con inclinación de la cabeza hacia la hemiespalda homolateral, y un pequeño estrechamiento de la hendidura palpebral homolateral al aducir, pueden completar el cuadro.
El síndrome puede ser consecutivo a un traumatismo, pero a veces es congénito. Los casos congénitos puede ser genéticos, al menos aquellos en que se ha comprobado un carácter familiar, pero las más de las veces son esporádicos sin una mutación reconocida (116,139).
BROWN-MARIE (SÍNDROME DE)
Es una ataxia hereditaria caracterizada por movimientos coreiformes, paresia piramidal, dislalia y deterioro mental progresivo (19,29,84). En el aparato ocular puede aparecer estrabismo, oftalmoplejía, nystagmus, anisocoria, y atrofia óptica.
Se han descrito casos esporádicos, pero en otros se detecta una clara transmisión hereditaria de tipo autosómico, generalmente recesivo, pero a veces dominante.
CFEOM (SÍNDROMES)
Vide: Fibrosis congénita de los músculos oculomotores (Síndrome de).
CHEDIAK-HIGASHI (SÍNDROME DE)
El síndrome de Chediak-Higashi (36,92) es un raro síndrome caracterizado por inclusiones citoplásmicas en los leucocitos. Añade anemia, neutropenia, trombocitopenia y albinismo óculo-cutáneo, y frecuentemente, se acompaña en el aparato oculomotor de nystagmus. Suele llevar a una muerte temprana.
El síndrome es de transmisión recesiva autosómica. Suele aparecer en hermanos albinos nacidos de padres consanguíneos. El gen responsable de este síndrome, ya identificado, se ha denominado CHS-1 o LYST, pues codifica la proteina LYST (101).
COCKAYNE (SÍNDROME DE)
Este síndrome, descrito en 1936 (39), asocia enanismo con atrofia retiniana y sordera, a lo que a veces se añaden problemas oculomotores internos (mala dilatatación pupilar con midriáticos) o externos (enoftalmos, heteroforias). Suele empezar a manifestarse tras los años de lactancia.
Su herencia es autosómica recesiva. Civantos (38,189) observó un caso con 47 cromosomas, por una trisomía del cromosoma 20.
COGAN (SÍNDROME DE)
Desde que Cogan (40) describiera hace más de medio siglo la apraxia oculomotora congénita con movimientos cefálicos espasmódicos, se han descrito numerosas variantes La forma más frecuente es una apraxia congénita del tercer par craneal, con imposibilidad de mover voluntariamente los ojos horizontalmente. Entre las variantes descritas está la de parálisis conjugada del motor ocular común. Se observa en niños, que generalmente mejoran con el tiempo (111).
Se han publicado casos consecutivos a traumatismo obstétrico, pero en algunos pocos hay un factor hereditario (178), de transmisión autonómica recesiva, con predominancia masculina. El gen responsable de las formas hereditarias, aún no identificado, parece estar situado en el brazo largo del cromosoma 2, locus 2q13.
CORNELIA DE LANGE (SÍNDROME DE)
Descrito por la Dra. de Lange en 1933 (45) y por Brachmann en 1916 (27), se caracteriza este síndrome por un desorden del desarrollo pre- y postnatal que se manifiesta por anormalidades de cabeza, tronco y extremidades (microcefalia, orejas bajas, dientes separados, hirsutismo) y cerebrales (disgenesia, menor número de neuronas, retraso psicomotor). En el aparato ocular hay cejas arqueadas, pestañas largas, y a menudo, estrabismo.
Su prevalencia es de 1x20.000 neonatos. Aproximadamente el 99% de los casos son esporádicos. Los casos hereditarios suelen ser autosómicos dominantes. El defecto radica en el gen NIPBL que produce una proteína, aún desconocida pero denominada delangina, cuya función parece ser regular las cromátidas. Un fenotipo similar al síndrome de Conelia de Lange parece observarse en pacientes con una duplicación de la banda q26-27 del cromosoma 3.
CRI DU CHAT (SÍNDROME DEL)
El síndrome del «cri du chat» (maullido de gato) (109), conocido por su nombre en francés en toda la literatura científica, se denomina así por la similitud del llanto de los lactantes con el miau del gato. Se manifiesta ya durante los primeros meses de vida por laringe y epiglotis pequeñas, microcefalia discreta, anomalías dentales, bajo peso al nacer, hipotonía muscular, retardo mental y disgenesia cardíaca. Hacia la tercera o cuarta década de la vida suele haber canicie incipiente. En el aparato ocular puede haber estrabismo generalmente divergente, hipertelorismo ocular, epicanthus y hendiduras palpebrales antimongoloides.
Es un raro síndrome que se da en 1 de cada 40.000 nacimientos. Se transmite de modo autosómico dominante. Está causado por la deleción parcial del brazo corto del cromosoma 5. Suele haber pérdida de varios genes, pero la zona crítica está en la banda 5p15.2. Aparece algo más en niñas, según la relación hembra/varón de 2/1, y se piensa que esto puede deberse a una mayor mortalidad masculina.
CROMOSOMA 21 TRIPLE (SÍNDROME DEL)
Vide: Down (Síndrome de).
CROMOSOMA X FRÁGIL (SÍNDROME DEL)
También conocido como síndrome de Martin-Bell (122), es la primera causa hereditaria de retraso mental. Se caracteriza por hiperactividad con déficit de atención, convulsiones que suelen desaparecer en la adolescencia, pies planos por laxitud articular y prolapso de válvula mitral. En el aparato ocular es característico el estrabismo.
Se debe este síndrome a una repetición de una secuencia del ADN del cromosoma X, el gen FMR1, que repite el trinucleótido CGG (Citosina-Guanina-Guanina). Cuando este trinucleótido se repite más de 200 veces, este gen se inactiva, es decir, deja de expresarse, produciendo un síndrome del cromosoma X frágil.
CROUZON (SÍNDROME DE)
El síndrome de Crouzon (43) se manifiesta por disóstosis craneofacial. con sinostosis prematura de las suturas craneales, oxicefalia y órbitas cortas, nariz en pico de loro e hipoplasia maxilar superior. En el aparato ocular es frecuente el estrabismo divergente, nystagmus, hipertelorismo y exoftalmos, ojo seco tantálico por exposición corneal, y hendiduras palpebrales antimongoloides. La afectación del nervio óptico es rara (171).
La enfermedad es de herencia autosómica dominante. Sus manifestaciones son ya notorias en el momento del nacimiento.
DAF (SÍNDROME)
El síndrome DAF (Downgaze palsy, Ataxia-athetosis, Foamy macrophages) (115) se considera como la variante C del síndrome de Niemann-Pick (Vide síndrome de Niemann-Pick). De las 5 variantes actualmente admitidas, ésta es la única que asocia un problema de la motilidad ocular, debido a un trastorno mesencefálico supranuclear oculomotor que dificulta y acaba impidiendo la mirada voluntaria vertical hacia abajo. Más tarde se afectan también los centros supranucleares ponto-mesencefálicos de la movilidad ocular horizontal. Los reflejos vestíbulo-oculares quedan mantenidos. La muerte suele sobrevenir a los 5-15 años, pero a veces se llega a la edad adulta
Esta forma C de síndrome de Niemann-Pick se debe en el 95% de los casos a una mutación del gen NPC1 localizado en el cromosoma 18, locus 18q11-q12. Este gen codifica una glicoproteína que participa en el transporte intracelular del colesterol hacia la membrana celular y retículo endoplásmico, en las células gliales cerebrales próximas a las tenminaciones sinápticas. El otro 5% de los casos se debe a una mutación del gen NPC2, localizado en el cromosoma 14, locus 14q24.3, que codifica una proteína lisosomal que se une al colesterol.
DE MORSIER (SÍNDROME DE)
El syndrome de De Morsier (46) o displasia septo-óptica incluye un desarrollo anormal de la hipófisis y septum pellucidum (parte del cerebro que separa ventriculos lateral del los cuernos cerebrales). Se añade trastornos olfativos y genitales. En el aparato visual puede haber hipoplasia de los nervios ópticos y estrabismo.
Los casos en que se ha determinado una transmisión hereditaria, ésta ha sido autosómica recesiva. Recientemente se ha encontrado que se debe a una mutación del gen HESX1 localizado en 3p21.2-21.1.
DELECIÓN PARCIAL DEL CROMOSOMA 18 (SÍNDROME DE)
Este síndrome, descrito primeramente por Grouchy (88) en 1964, asocia como anomalías más frecuentes enanismo, displasia hemifacial, retardo mental y autismo. Entre las manifestaciones oculares, están el estrabismo, nystagmus horizontal, hendiduras palpebral oblicuas, epicanthus y pupilas ovales.
La causa es la deleción de parte del brazo largo del cromosoma 18. A veces se trata de una translocación (3q, 18q, u otras) con pérdida o no de parte del cromosoma. En el 75% de los casos no hay translocación en los padres.
DISPLASIA OSTEO-METAFÍSEA (SÍNDROME DE)
La displasia osteo-metaphysea es un síndrome descrito por Golden (78) (1977), que afecta principalmente al sistema óseo (enanismo, puente nasal ancho y plano, narinas antevertidas, pectus carinatum, cuerpos vertebrales aplastados, cifosis, deficiente osificación de las metáfisis y epífisis, hiperextensibilidad de las articulaciones), y cuyo cuadro ocular consta de estrabismo, nystagmus e hipertelorismo. A veces se han descrito formas hereditarias.
DISTIQUIASIS-LINFEDEMA (SÍNDROME DE)
Descrito por primera vez en 1964 (59), se caracteriza por distiquiasis desde el nacimiento y veces raspado surfocular, y pocos años después, por linfedema de las piernas y pterygium colli.
Es de herencia autosómica dominante. Se debe a una mutación del gen FOXC2 6 en 16q24 (28).
DISTONÍA TORSIONAL (SÍNDROME DE)
Consiste en distonía torsional y parpadeo espasmódico (107), del que últimamente se han citado casos con problemas oculomotores. Se da preferentemente en Filipinas, especialmente en la isla de Panay.
Se trasmite de modo recesivo ligado al sexo (130).
DOWN (SÍNDROME DE)
El síndrome de Down (49) se caracteriza por mongolismo e idiocia, a lo que se añaden algunas anormalidades esqueléticas, pliegue palmar transversal, hiperextensibilidad de articulaciones, hipersalivación, etc. En el aparato ocular no es raro el estrabismo y/o el nystagmus, hendiduras palpebrales pequeñas y epicánthicas, hipertelorismo, hipoplasia de iris, miopía y catarata. En la revisión de Navarro et al. (134) de 195 niños con síndrome de Down encontraron ametropías en el 64%, estrabismos en el 52%, nystagmus en el 21%, y cataratas en el 10%. En el estudio de Galán et al. (72) el 43% de los niños de síndrome de Down tenía estrabismo.
La causa está en una trisomía del cromosoma 21, que a su vez puede estar en relación con factores dependientes de la edad parental.
DUANE (SÍNDROME DE)
Vide: Stilling-Türk-Duane (Síndrome de).
DUCHENNE (SÍNDROME MIOPÁTICO DE)
La miopatía de Duchenne (72) es una debilidad de la musculatura estriada que se inicia en los primeros años de la vida, e impide la marcha ya antes de la pubertad. También puede afectarse la musculatura lisa.
Se produce por una mutación del gen DMD que codifica la dystrophina, y que se localiza en Xp21.2. Se transmite de modo recesivo ligado al cromosoma X, afectando principalmente a los varones. Las mujeres conductoras pueden expresar algunas de las manifestaciones del síndrome según las características del alelo sano.
EHLERS-DANLOS (SÍNDROME DE)
Es una fibrodisplasia elástica generalizada por hipoplasia de la colágena (54,44), con piel atrófica e hiperelástica, hiperlaxitud articular y pseudoxantomas. En los ojos puede haber hipotonía muscular extrínseca con blefarochalasis (signo de Méténier) (124), estrabismo, escleras azules Se presenta ya desde el nacimiento.
Es de transmisión hereditaria, de tipo autosómico dominante, regular o irregular, y con penetrancia relativamente baja.
ELLIS-van CREVELD (SÍNDROME DE)
Displasia condro-ectodérmica descrita en 1940 por Ellis y van Creveld (55), caracterizada por extremidades cortas con genu valgum, pies zambos, polidactilia, distrofia de uñas, anomalías dentales y a veces cardiopatía y retardo mental. En los ojos puede haber estrabismo convergente, coloboma de iris y catarata congénita,
Se hereda de forma autosómica recesiva. En el 25% de los pacientes, sus padres son consanguíneos.
ESCLEROSIS EN PLACAS O ESCLEROSIS MÚLTIPLE
Esta enfermedad, descrita por Charcot (35) en 1868, se caracterizada por la aparición de placas mielínicas y áreas de desmielinización alrededor de los axones del sistema nervioso central. Las placas aparecen por brotes. La edad de comienzo más frecuente es entre los 20 y los 40 años, siendo rara antes de los 10 años o después de los 50 años. Se caracteriza por falta de coordinación y equilibrio, debilidad, dificultad para andar y hablar, vértigo y en el aparato ocular, puede haber parálisis oculomotoras de origen nuclear y supranuclear (oftalmoplejia internuclear) y neuropatía óptica retrobulbar.
Su frecuencia varía según la zona del mundo, siendo más frecuente en la EEUU y Europa Occidental. En España, Alcalá et al. (1993) le dan una prevalencia de 1 de cada 2.500 personas. La enfermedad es más frecuente en mujeres que en hombres.
La esclerosis en placas es una enfermedad autoinmune que ataca la mielina, cuya causa genética no está bien establecida. Cuando en dos gemelos homozigotes uno tiene esclerosis en placas, el otro tiene un 30% de probabilidades de desarrollarla también.
Entre los pacientes de esclerosis en placas se encuentra con más frecuencia que entre la población general el haplotipo HLA DRB1 del sistema mayor de histocompatibilidad de clase II. Otro haplotipo también más frecuente es el HLA DR13. Como candidatos a provocar esclerosis en placas se ha señalado, sin confirmación posterior, la proteína CD45 cuyo gen está en 1q31-q32, y la apolipoproteína E, cuyo gen está en 19p13.2.
FIBROSIS CONGÉNITA DE LOS MÚSCULOS OCULOMOTORES (CFEOM) (SÍNDROME DE)
Los síndromes CFEOM (Congenital Fibrosis of the Extra Ocular Muscles) tienen en sus distintas variantes una prevalencia que se calcula en 1 por 230.00 habitantes. En el mundo se calcula que hay 20.000-25.000 casos, y en España 150-200 casos. En España han sido publicados casos familiares y no familiares por Hidalgo (91), Villalonga (181), Perea (139) y otros.
Se han detectado hasta el presente tres formas distintas de CFEOMs:
El CFEOM-1 tiene su trastorno localizado el gen KIF21A de la región centromérica del brazo largo del cromosoma 12 (56), en 12p11.2-q12. Este gen expresa una kinesina responsable del transporte intracelular asociado a los microtubuli, y que en las neuronas está implicada en el transporte anterógrado. La tara se suele transmitir de forma autosómica dominante (137). La patogenia del síndrome es neurogénica y no biogénica, y la lesión se ha localizado en el núcleo del nervio motor ocular común. Las anomalías musculares parecen ser secundarias. Se han visto casos asociados al síndrome de Marcus Gunn (62) y al de Möbius (137).
El paciente sufre una limitación de los movimientos oculares verticales. En reposo los ojos se colocan en hipotropía, y no pueden elevarse sobre la horizontal. Los movimientos horizontales pueden persistir, o estar también bloqueados. Se puede añadir esotropía o exotropía. Se añade blefaroptosis. El paciente debe mirar con tortícolis de extensión.
El CFEOM-2 resulta de la mutación del gen ARIX, situado en el brazo largo del cromosoma 11, en 11q13. Se transmite de forma recesiva autonómica (133). La mutación provoca la ausencia de los pares craneales III y IV, así como del locus coeruleus y de otros núcleos neuronales.
El cuadro clínico se manifiesta por una oftalmoplegia total, a veces con ligera conservación de la abducción. La blefaroptosis es bilateral y muy marcada.
El CFEOM-3 tiene el gen causal en el brazo largo del cromosoma 16, en 16q24.2-q24.3 (48,56). Se transmite de forma autosómica dominante. Sus manifestaciones clínicas son similares a las del síndrome CFEOM-1, con ligeras variantes, como una menor blefaroptosis.
FORSIUS-ERICKSON (SÍNDROME DE)
También conocido como síndrome de la isla Aaland por ser allí relativamente frecuente, el síndrome de Forsius-Erickson (64) es un trastorno familiar cuyas manifestaciones más características son oculares: albinismo incompleto del fundus oculi, degeneración tapeto-retiniana, displasia macular, protanopía, nystagmus, y frecuentemente, estrabismo y miopía. A menudo se asocian trastornos neurológicos y auditivos.
El síndrome afecta plenamente sólo a varones, mientras que las hembras, o tienen un fenotipo normal, o padecen sólo nystagmus latente muy discreto. Según van Vliet et al. (177) la transmisión es dominante ligada al cromosoma X, con escasa penetración en las hembras heterozigotes, o bien recesiva intermediaria ligada al cromosoma X, capaz de provocar un ligero nystagmus en las portadoras.
FRANCESCHETTI (SÍNDROME DE)
El síndrome de Franceschetti (65,67) es una disóstosis maxilofacial aparecida ya en la vida fetal, con hipoplasia cigomática, malformaciones dentales, párpados con fisura antimongoloide, ausencia de puntos lacrimales y de glándulas de Meibomio, a veces con microftalmía, coloboma de iris y trastornos oculomotores externos.
En España, datos del del ECEMC (Estudio Colaborativo Español de Malformaciones Congénitas) estiman una frecuencia de 0,07 por cada 10.000 nacimientos.
Se transmite de forma autosómica dominante irregular, con penetrancia incompleta y expresividad variable. El responsable del síndrome es la alteración del gen TCOF1, localizado en el brazo largo del cromosoma 5 (5q32-q33.1), de la que se han descrito más de 30 mutaciones Se han citado casos asociados a translocación t(5;13)(q11;p11) (4) con descenso significativo de hexosaminidasa B.
FRANÇOIS-HALLERMANN-STREIFF (SÍNDROME DE)
Vide: Hallermann-Streiff (Síndrome de).
FREEMAN-SHELDON (SÍNDROME DE)
Es una displasia cráneo-carpotarsiana, también conocida como síndrome de la cara de silbido (69), en el que puede añadirse blefarofimosis, enoftalmos y estrabismo convergente. Es muy raro.
Posible se hereda en forma autosómica dominante. Temtamy (168) 1966, presentó dos generaciones en tres familias.
FRIEDREICH (SÍNDROME DE)
Vide: Ataxia hereditaria de Friedreich.
GANGLIOSIDOSIS GM1
Es una gangliosidosis generalizada en la que por falta de gangliosido-b-galactosidasa se acumulan los gangliósidos GM1 en los tejidos nerviosos hepático, esplénico y renal. En fondo de ojo es muy característico, pues la foveola se ve de color rojo cereza rodeada de un anillo perifoveolar blanquecino. Los trastornos oculomotores no son excepcionales. Se empieza a manifestar a los días o semanas de nacer, y raramente el paciente sobrevive a los tres años. En la forma de aparición tardía se pueden alcanzar los 15 años de edad.
Se transmite según el modo autosómico recesivo (47,147). El gen del enzima causal está localizado en 3p21.33.
GANGLIOSIDOSIS GM2
Vide: Tay-Sachs (Síndrome de) y Sandhoff (Enfermedad de).
GARCÍA-DRUMMOND (SÍNDROME DE)
Reúne este síndrome (73,50) hipercalcemia asociada a trastornos intestinales, vómitos, estreñimiento y a veces enanismo y osteosclerosis. Como afectaciones oftalmológicas se han citado estrabismo, nystagmus y epicanthus. Suele empezar a manifestarse en la infancia (77).
El síndrome se hereda de forma recesiva.
GAUCHER (SÍNDROME O ENFERMEDAD DE)
La llamada enfermedad de Gaucher (76) es un déficit enzimático que provoca acumulación de glucocerebrósidos en diversos tejidos del organismo. Se manifiesta por alteraciones hepáticas, hematológicas, óseas, pulmonares y neurológicas, entre los que destacan las parálisis supranucleares. Suele empezar a manifestarse poco después del nacimiento y a menudo termina con la vida en pocos años. Se han establecido 3 formas clínicas: el tipo I no tiene síntomas neurológicos ni oculomotores, y los tipos II y III, sí.
Es de trasmisión autosómica recesiva. Se debe a la mutación del gen de la b-glucocerebrosidasa, localizado en1q21.
GOLDENHAR (SÍNDROME DE)
Descrito hace más de medio siglo por Goldenhar (79), reúne anomalías vertebrales, apéndices auriculares y fístulas preauriculares, hipoplasias faciales, y en un 10% de los casos, retardo mental. En el aparato ocular se manifiesta por quistes dermoides epibulbares, generalmente en cuadrante témporo-superior del limbo esclerocorneal; fisuras palpebrales antimongoloides, y frecuentemente estrabismo, blefarofimosis, y blefaroptosis con o sin lagoftalmo. Se da en 1 caso por cada 5.000-20.000 neonatos. Afecta más a varones que a hembras, en proporción 3:2.
El 98% de los casos son esporádicos. El 2% restante sigue una herencia autosómica dominante (148).
GOLTZ (SÍNDROME DE)
Su manifestación más evidente, a menudo ya visible al nacimiento, es una hipoplasia dérmica focal con telangiectasias cutáneas y papilomas en las mucosas, hernia inguinal y umbilical. En el aparato ocular es frecuente el estrabismo, así como el coloboma irídico y coroideo (80).
Casi siempre afecta a hembras. Se ha señalado una transmisión ligada al cromosoma X, de tipo dominante, con letalidad en los varones (155,172).
GORLIN-GOLTZ (SÍNDROME DE)
Su carácter más distintivo es la presencia de numerosos nevi basales preferentemente en párpados y cara y de anomalías de costillas. En ojos puede haber estrabismo convergente, coloboma coroideo, ensilladura nasal ancha y reborde supraorbital prominente.
Aunque el síndrome ya había sido descrito por Jarisch en 1894 (97), ha recibido el nombre de Gorlin-Goltz (85) porque en 1960 estos autores lo describieron extensamente, y detectaron casos hereditarios, para los que sugirieron una herencia autosómica dominante, de escasa penetrancia.
GREENFIELD (SÍNDROME DE)
Síndrome de Greenfield (86) es el más extendido de los diversos nombres que recibe una leucodistrofia metacromática infanto-juvenil, caracterizada por la desmielinización de fibras nerviosas que aún no están completamente mielinizadas. Se manifiesta en forma de ataxia, demencia, blefaroptosis y estrabismo. Puede llevar a la sordera y a la ceguera por atrofia del cortex acústico y visual.
Se han descrito formas de transmisión autonómica recesiva. En formas de presentación en edad adulta se han señalado casos ligados al sexo, de transmisión dominante.
GREIG (SÍNDROME DE)
Su manifestación más característica es el hipertelorismo ocular (87), a lo que se añade ensilladura nasal ancha y aplanada, y a menudo, labio leporino y paladar ojival. En los ojos es frecuente la presencia de parálisis bilateral del sexto par, epicanthus, y deformaciones de párpados y cejas. En casos de hipertelorismo extremo falta la fusión binocular y a veces hay atrofia del nervio óptico por tracción mecánica.
El síndrome se describe generalmente como esporádico, pero Bojlen et al. (23) publicaron el genodendro de una familia con 24 casos a lo largo de 4 generaciones, con transmisión autosómica dominante. Y Fuller et al. (71) citaron una familia con dos hermanos de distinto sexo ambos afectos, la mujer con una trisomía gonosómica XXX, y el varón con una tetragonosomía XXXY.
HALLERMANN-STREIFF (SÍNDROME DE)
Este síndrome (90,165), también conocido como de «cabeza de pájaro», se caracteriza por malformaciones craneofaciales en forma de braquicefalia, anomalías dentales, cabello escaso y ligero retardo mental. A veces, se añade osteosclerosis, hiperextensibilidad articular y enanismo. En el aparato ocular puede haber estrabismo y nystagmus, y disminución de visión por cataratas congénitas o por atrofia retiniana.
Es frecuente la afectación familiar en pareja de progenitores consanguíneos. Se da tanto en varones como en hembras.
HUNTINGTON (ENFERMEDAD O COREA DE)
Trastorno degenerativo del sistema nervioso central (93) que suele comenzar a manifestarse pasada la pubertad, progresa lentamente por movimientos coréicos (en griego, coroV,chorós = danza) y demencia, llevando a la muerte a edades medias de la vida. Las manifestaciones oftalmológicas son principalmente oculo-motoras, como inestabilidad de la fijación y saccadas horizontales apráxicas, que más tarde afectan también a los movimientos verticales.
Afecta a 1 de cada 15.000 personas. Se transmite según el modo autonómico dominante. Se debe a la mutación del gen IT15 (Important Transcript 15) localizado en el cromosoma 4, en 4p16.3, y las manifestaciones clínicas varían grandemente según el tamaño y actividad del alelo sano.
HURLER (SÍNDROME DE)
La mucopolisacaridosis de Hurler (94) es un trastorno metabólico que se manifiesta por marcado retraso del desarrollo, dentición tardía, infantilismo, cifosis dorsolumbar, espina bífida, hepatomegalia y esplenomegalia. En los ojos puede haber lesiones surfoculares corneales (opacidad difusa por infiltración lípida subepitelial, a veces con ausencia de membrana de Bowman), estrabismo convergente, hipertelorismo, y degeneración retiniana. La muerte suele sobrevenir entre la pubertad y el inicio de la adultez.
Hay casos esporádicos, y casos familiares de herencia autonómica recesiva.
INCONTINENTIA PIGMENTI (SÍNDROME DE)
Vide: Bloch-Sulzberger (Síndrome de).
JACOBS (SÍNDROME DE)
El síndrome de Jacobs (96) o de trisomía X es frecuentemente asintomático, pero cuando no es así, se puede manifestar por dismenorrea, y ligeras dismorfias bucales como paladar ojival y dentición anormal. En el aparato ocular puede haber estrabismo, epicanthus, hendidura palpebral mongoloide e hipertelorismo.
El síndrome se produce en mujeres que han nacido con un cromosoma supernumerario X, lo que parece ocurrir en aproximadamente una niña por cada mil nacidas. A veces se añade también una trisomía autosómica. Las mujeres con síndrome XXX son fecundas y dan hijos normales.
JOUBERT (SÍNDROME DE)
Se trata de una dismorfia congénita (99) producida por hipoplasia vermo-cerebelosa y anomalías ponto-mesencefálicas, con retardo psicomotor, hipotonía y ataxia. En el aspecto externo, destacan frente prominente y orejas bajas. En el aparato ocular puede haber trastornos de la motilidad ocular vertical y horizontal, distrofia corio-retiniana, hipoplasia del nervio óptico con pérdida de visión, implantación alta de las cejas y epicanthus.
Se transmite según el modo autosómico recesivo. Se han descrito varios genes afectados localizados en los cromosomas 6, 9, 11 y 17, respectivamente en los loci 6q23.3, 9q34.3, 11p12-q13.3 y 17p11.2. En algunos niños con síndrome de Jouvet se ha encontrado el gen mutado de la nefronoptisia NPHP1 localizado en 2q13, el cual además de actuar en el desarrollo renal puede participar en el del cerebelo y tronco cerebral.
KEARNS-SAYRE (SÍNDROME DE)
El síndrome de Kearns-Sayre (100) es un trastorno multisistémico que suele empezar a manifestarse antes de los 20 años, y se caracteriza por una oftalmoplejía externa progresiva, con debilidad muscular esquelética, retinitis pigmentosa, disminución de audición, defectos cerebelosos, y trastornos endocrinos como diabetes mellitus, hipotiroidismo e hipogonadismo, todo ello influyendo en una baja estatura.
Es una enfermedad que se transmite por el ADN mitocondrial, por lo que se hereda a través de la madre, pero la padecen tanto los varones como las hembras (30).
KLEIN-WAARDENBURG (SÍNDROME DE)
Esta displasia interoculo-irido-dermato-auditiva (103,185) se manifiesta por raíz nasal ancha y sin ángulo naso-frontal, áreas de albinismo (mechón blanco frontal) y de hiperpigmentación, y sordera o sordomudez. En los ojos puede haber estrabismo convergente, hipertelorismo ocular con agrandamiento del mesofrión e hiperplasia de la porción medial de las cejas, hipoplasia de carúnculas, y heterocromía iridis o aniridia. En las razas caucásicas afecta a 1 de cada 40.000 personas.
Actualmente se admiten de momento 4 tipos de Klein-Waardenburg, de los que el I, II y III son de transmisión autosómica dominante con expresividad variable, y el tipo IV, que es el más ligado a albinismo, autosómica recesiva (53,130). También en algunos genodendros se ha detectado un gen presuntamente ligado al cromosoma X.
KOHN-ROMANO (SÍNDROME DE)
Como ya indica el título del artículo de los autores que dieron nombre a este síndrome (104), las manifestaciones externas más notorias son blefaroptosis, blefarofimosis, epicanthus inverso y telecanthus. A ello no es raro que se asocien alteraciones oculomotoras, como estrabismo convergente o divergente, limitación de la acción de los rectos superiores o hiperacción de los oblicuos inferiores. También puede haber disgenesia de los puntos lacrimales.
El síndrome es de trasmisión autosómica dominante, con una penetrancia del 100%.
LAURENCE-MOON-BARDET-BIEDL (SÍNDROME DE)
El síndrome (106,16) reúne un cuadro de retinitis pigmentosa con disfunción de fotorreceptores, obesidad y trastornos óseos (escasa estatura, genu valgum, polidactilia, turricefalia), neurológicos (abiotrofia de la región diencéfalo-hipofisaria, movimientos coreiformes) y endocrinos (hipogenitalismo). En el aparato ocular, aparte de la disminución de visión escotópica defectuosa propia de la retinitis pigmentaosa, es frecuente el nystagmus, la oftalmoplejia externa, el coloboma de iris, la blefaroptosis y el epicanthus. Comienza en la infancia, y no acorta la vida. Como en todos los síndromes descritos en los siglos XIX y XX, basados fundamentalmente en cuadros clínicos, este síndrome podría incluir diversas variantes derivadas de distintas variaciones genómica en vías de determinación.
Se han descrito diversas manifestaciones del síndrome con un gen de transmisión dominante autosómica, de un gen recesivo autosómico y de un gen recesivo ligado al sexo.
LEBER (SÍNDROME DE)
Más conocido como enfermedad de Leber o neuropathia optica familiaris Leberi, es una enfermedad que se manifiesta en o tras la pubertad y se caracteriza por la aparición de una optoneuritis retrobulbar bilateral aguda, generalmente indolora, que tras fases de mejoría y empeoramiento, acaba llevando en unos años a la ceguera legal, y a veces a la ceguera total. Se pueden asociar otras manifestaciones, como hiperreflexia tendinosa, epilepsia, trastornos piramidales y sordera. Entre estas manifestaciones se ha descrito, aunque muy raramente, la paraplejia espasmódica de Strümpell y las afectaciones de la motilidad ocular extrínseca.
Se da en uno de cada 50.000 personas. El hombre no transmite la enfermedad a su descendencia, pero la mujer, manifieste o no manifieste la enfermedad, la transmite a todos sus hijos (149).
Para explicar que los varones no transmiten la enfermedad, se han buscado 3 explicaciones: 1) La forma más frecuente es la de transmisión por el ADN mitocondrial materno, y no por herencia cromosómica nuclear (95). 2) Los espermatozoides transmisores del cromosoma tarado producen zigotos no viables. 3) En el varón afecto se produce una mutación inversa que normaliza el gen tarado (83). El 80% de las mutaciones codificadas corresponden a 11778A, 3460A y 14484C.
Cuando la transmisión de la enfermedad de Leber es autosómica suele seguir un patrón recesivo ligado al cromosoma X, si bien un reducido número de genodendros muestran una herencia autosómica dominante (130).
LEIGH (SÍNDROME DE)
El síndrome de Leigh (108) o encefalo-mielopatía necrosante subaguda es una necrosis encefálica que se manifiesta desde las primeras semanas de vida por ataxia cerebelosa, síndrome piramidal, retardo psicomotor y crisis comiciales. En el aparato oculomotor puede incluir parálisis horizontal de la mirada, oftalmoplejías internucleares y sacudidas nystágmicas.
Se produce por diversos genes: La forma debida al gen SURF-1, localizado en 9q34.3, es de herencia autosómica recesiva. La forma debida al gen de la flavoproteína se localiza en 5p15. La forma debida al gen CPDH,E1a, localizada en el cromosoma X, locus Xp22.2-p22.1 es de herencia recesiva ligada al sexo. Hay también una forma mitocondrial de la subunidad 5 del complejo I. Las formas debidas a los genes NDUFS1, NDUFS4 y NDUFS7 no han sido aún bien localizadas.
LOUIS-BAR (SÍNDROME DE)
Descrito por la doctora belga Denise Louis-Bar (113) en 1941, el síndrome de ataxia-telangiectasia asocia una cuadro neurológico central (ataxia, abolición de reflejos tendinosos, corea, retardo mental) con un cuadro ocular de telangiectasias conjuntivales y trastornos oculomotores (estrabismo en el 30% de los casos, apraxia similar a la del síndrome de Cogan) . Es frecuente que se añada un déficit inmunitario, y propensión a desarrollar linfomas.
Transmisión autosómica recesiva. El gen tarado es el ATM, formado por 66 exones, y localizado en el cromosoma 11, en 11q23.3. Este gen codifica una proteinkinasa que regula la reparación de los filamentos de ADN.
LOWE (SÍNDROME DE)
También conocido este síndrome (114) como síndrome oculo-cerebro-renal, asocia acidosis generalizada retardo mental, alteraciones renales, osteomalacia, y en el aparato ocular glaucoma congénito, nystagmus y miosis.
Todos los casos descritos corresponden a varones, por lo que se ha pensado que se deba a un gen ligado a un gonosoma, de características recesivas. Recientemente se ha identificado herencia ligada al gonosoma X, por alteración del gen OCRL1, que expresa la proteína enzimática fosfatidilinositol-4,5-bifosfato, que interviene en el transporte intracelular (75).
MARCUS GUNN (SINDROME DE)
El síndrome de Marcus Gunn (89), también conocido como jaw winking phenomenon, es una sincinesia mandíbulo-palpebral en la que un ojo con blefaroptosis unilateral de un párpado superior, se eleva rápidamente al abrir la boca o al masticar. El ojo izquierdo se afecta con más frecuencia.
El origen parece ser central, debido a la existencia de fibras aberrantes entre el núcleo elevador del párpado superior y el núcleo de la rama motora del trigémino (músculo pterigoideo externo). Recientemente se ha descrito un caso asociado a un síndrome de fibrosis congénita de músculos oculomotores CFEOM-1 (62).
El carácter hereditario es raro, pero cuando se ha podido demostrar, éste ha sido de modo autosómico dominante irregular (63,110,125).
MARFAN (SÍNDROME DE)
Es una distrofia mesodérmica congénita descrita en 1986 por Marfan (118), que se manifiesta por trastornos esqueléticos (tórax asimétrico, aracnodactilia, dolicocefalia, ligamentos relajados). En los ojos es frecuente la ametropía (miopía, hipermetropía o astigmatismo), estrabismo divergente, nystagmus y la subluxación del cristalino con esferofaquia.
Se ha descrito la transmisión autosómica dominante y la autosómica recesiva. La enfermedad se debe a una mutación del gen de la fibrilina, que es uno de los componentes de las microfibrillas de la matriz extracelular. Este gen se localiza en el cromosoma 15.
MARIE (ATAXIA CEREBELOSA DE PIERRE MARIE)
Vide: Ataxia espino-cerebelosa hereditaria de Marie.
MARÍN AMAT (SÍNDROME DE)
El síndrome de Marín Amat (120,121) consiste en la mayor caída de un párpado superior ptótico cuando se abre la boca ampliamente, fenómeno que algunos autores han denominado posteriormente «fenómeno de Marcus Gunn inverso». Se ha encontrado tras parálisis faciales periféricas, y se ha supuesto como en el síndrome de Marcus Gunn también la anastomosis entre el V y VII par.
El autor lo describió como consecutivo a traumas, pero posteriormente se han descrito casos en que se sospecha un factor hereditario, sin haberse demostrado.
MAULLIDO DE GATO (SÍNDROME DEL)
Vide: Cri du chat (Síndrome del).
MIASTENIA (SÍNDROMES DE)
Las miastenias se deben a trastornos de la unión sináptica neuromuscular, una compleja estructura que puede alterarse por diversos genes mutados.
La miastenia neonatal pasajera se piensa puede ser por el paso diaplacentario de la madre al hijo de anticuerpos anti-receptores a la acetilcolina (Ac-ARACh). Las miastenias congénitas persistentes se dividen en 3 grupos: el grupo I incluye los casos hereditarios por alteraciones de los receptores de la acetilcolina o por déficits de colinesterasa; el grupo II incluye los casos hereditarios por tratornos postsinápticos; y el grupo III, los casos esporádicos. Todas ellas producen debilidad muscular que se inicia ya en la lactancia, edad infantil o pubertad, y que aumenta a lo largo del día. En el aparato ocular se manifiesta por blefaroptosis, paresias oculomotoras o estrabismo. Los primeros músculos implicados suelen ser el elevador del párpado superior y los rectos interno, inferior y superior. El orbicular raramente se afecta, y la musculatura intrínseca, nunca.
Los genodendros de miastenias del grupo I muestran una transmisión autosómica recesiva. En este grupo I se han detectado mutaciones de los genes F1MG1, localizado en la banda 17p13, o del gen CHAT (F1MG2), localizado en 10q11.2; del gen ColQ localizado en 3p24.2; en el gen de la rapsina (receptor associated protein of synapses) localizado en 11p11.2-p11.1. Los genodendros de las miastenias del grupo II son autosómicas dominantes. En este grupo II se han detectado mutaciones en el gen a-RACk, localizado en 2q24-q31, o en el b1RACh, localizado en 17p13.1; o en el den dRACh, localizado en 2q33-q34 o en 2q37.1. Las miastenias del grupo III son esporádicas.
MÖBIUS (SÍNDROME DE)
Es una diplejía facial congénita que afecta al VI y VII pares craneales por aplasia de sus núcleos metencefálicos. A veces se añaden trastornos vestibulares y sordera (127). Raramente se asocia a lágrimas de cocodrilo, es decir, producción lacrimal cuando se estimula la salivación al comer; esto se debe a una inervación anormal de fibras salivatorias derivadas al nervio lacrimal. La creatinuria, encontrada por algunos autores, no ha sido confirmada por Prieto-Díaz et al. (144).
La mayoría de los casos son esporádicos (139). Últimamente se han descrito varios casos familiares, afectando los genes MBS1, MBS2 y MBS3 . El primero de estos genes se ha localizado en la región 13q12.2-q13 del brazo largo del cromosoma 13. El segundo, en el brazo largo del cromosoma 3, en 3q21-q22. Otro, en el brazo largo del cromosoma 10, en la región 10q21.3-q22.1 (105,126). Es posible que algún síndrome de Moebius tenga alguna relación con el síndrome CFEOM-1 (137,270).
MORSIER (SÍNDROME DE)
Vide: De Morsier (Síndrome de).
NAEGELI (SÍNDROME DE)
El síndrome de Naegeli (132) es una forma de incontinentia pigmenti de Bloch-Sulzberger, pero sin dermatitis ampollosa. Desde los primeros años de la vida la pigmentación cutánea reticular se asocia a hiposudoración, queratosis palmo-plantar y anormalidades dentales. En el aparato ocular es frecuente el estrabismo y el nystagmus.
La herencia es autosómica dominante, afectando por igual a ambos sexos.
NEUROFIBROMATOSIS TIPO I
Vide: von Recklinghausen (Enfermedad de).
NIEMANN-PICK (SÍNDROME O ENFERMEDAD DE)
Es una esfingolipoidosis (135,140) caracterizada por acumulación de depósitos de esfingomielina en el retículo endoplásmico y de colesterol en las organelas intracelulares. La forma típica suele llevar a la muerte en los primeros años de vida. Según su sintomatología se ha descompuesto en 5 formas clínicas (A, B, C, D y E), de las que sólo la foma C asocia una parálisis oculomotora. Esta forma C era y suele seguir siendo conocida como síndrome DAF (vide: DAF, síndrome).
OKIHIRO (SÍNDROME DE)
El síndrome fue descrito por Temtamy (169) en 1975 como Duane-Radial Ray syndrome, y más tarde por Okihiro (136) en 1977. Se trata de una asociación de síndrome de Stilling-Türk-Duane con sordera, displasia ósea del radio y algunas anomalías menores como falta de dedo pulgar (139).
Se transmite según el modo dominante autosómico. Se trata de una mutación del gen SALL4, localizado en el cromosoma 20, en 20q13,13-q13.2, que juega un importante papel en el desarrollo del núcleo del VI par craneal.
PANTALLA DE ORDENADOR (SÍNDROME DE LA)
Desde hace más de un siglo se conoce la triple sinkinesia de visión cercana acomodación-convergencia-miosis. A principios de este siglo se vio que la sinkinesia es mucho más compleja, siendo las 3 principales asociaciones añadidas a este complejo la oculodepresión-blefarodepresión-cervicoflexión (131). Esto no ha empezado a manifestarse hasta la aparición de las pantallas de ordenador, en que por primera vez en la historia de la especie humana se pueden pasar horas con los ojos mirando de cerca en horizontalidad (en vez de en ínfero-convergencia). Esto obliga a contrarrestar la sinquinesia con una contractura cervical posterior para no flexionar el cuello, una contractura de oblicuos inferiores para no dirigir los globos oculares hacia abajo, y una contractura de elevador de párpado para no bajar el párpado superior. Estas contracturas producen cansancio, aumentan la abertura palpebral y disminuyen la frecuencia del parpadeo, lo que condiciona un síndrome de sequedad surfocular.
Muchos trastornos neurológicos de transmisión hereditaria o no hereditaria (158) dificultan las sinkinesias de visión cercana en inferoducción ocular (que es la forma fisiológica), así como su compensación en las recientes circunstancias de visión cercana en horizontalidad (lo que obliga a una compensación) manifestándose no sólo en ojo seco y contracturas cervicales, sino también en diplopía circunstancial.
PARÁLISIS DEL IV PAR (SÍNDROME DE)
La parálisis genética del nervio patético, aunque excepcional, puede existir. La incapacidad de inferoducción en adducción se suele compensar con un tortícolis con el mentón abajo y la cabeza inclinada hacia la hemiespalda opuesta al músculo paralizado, para permitir la fusión binocular.
Los casos familiares son excepcionales (26). Recientemente se ha encontrado en una familia una mutación del gen ARIX a nivel del exon 1 (98).
PELIZAEUS-MERZBACHER (ENFERMEDAD O SÍNDROME DE)
El síndrome de Pelizaeus-Merzbacher (138,123) (1885, 1910) es un trastorno difuso de la substancia blanca del sistema nervioso central. Comienza en la infancia. Comporta retraso físico, motor y mental: marcha inestable, temblor intencionzal, trastornos de la audición y del habla. En el aparato ocular hay un nystagmus pendular elipsoidal, asociado a un nystagmus vertical y a un balanceo de la cabeza. No es raro que este nystagmus mejore progresivamente. A veces se progresa hacia una atrofia del nervio óptico. Los cuadros clínicos se han distribuido en tres formas distintas (11).
Las tres formas del síndrome se transmiten en modo recesivo ligado al cromosoma X. El gen alterado se localiza en Xq22. Su misión es actuar sobre la proteolípido-proteína (PLP) que provoca y mantiene la mielinización de las fibras nerviosas. Este gen de la PLP está duplicado en 2/3 de los casos de niños afectos. El otro tercio de los casos se debe a deleciones parciales del gen, y da lugar a los casos más graves del síndrome (130).
En muy pocos casos, el síndrome es autosómico recesivo, y se debe a una mutación en el gen GJA12, localizado en 1q41-q42, gen que codifica la connexina, una proteína que se expresa en los oligodendrocitos.
PENTAGONOSOMÍA (SÍNDROME DE)
El síndrome Penta-X se manifiesta ya en las niñas afectas por hipogenitalismo y retraso mental, y en sus ojos, por miopía, epicanthus y estrabismo. Su cariotipo es 49-XXXXX. Las afectas tienen hasta 4 corpúsculos de Barr en las células del frotis conjuntival o bucal. La aneuplodia no es transmisible.
Otra pentagonosomía es el síndrome XXXXY. En los aproximadamente 50 casos publicados, la enfermedad no es heredable.
PIERRE MARIE
Vide: Marie
PRADER-WILLI (SÍNDROME DE)
Es un trastorno congénito y genético, que se manifiesta por obesidad, hipogonadismo, hipotonía muscular y retraso mental (142,143). El estrabismo no es frecuente. En España se han publicado 3 casos por Saá et al. (156), dos con esotropía y uno con exotropía.
Está causado por la deleción de un gen del cromosoma 15. En el 70% de los casos afecta al cromosoma 15 heredado del padre. En uno de los casos de Saá et al. se analizó el cariotipo, que mostró una deleción del brazo largo del cromosoma 15, afectando al banda 15q11-q12 .
PSEUDO-PARAHIPOTIROIDISMO (SÍNDROME DE)
Es una osteodistrofia hereditaria en la que las glándulas paratiroides funcionan normalmente, pero los riñones y el esqueleto no responden normalmente a la parathormona (3). La estatura es baja, el cuello corto, y hay tendencia a la obesidad. En el aparato ocular es frecuente el estrabismo, y menos frecuente las escleróticas azules y las leucofaquias cristalinianas puntiformes.
La herencia es autonómica dominante. La presentación es dos veces más frecuente en hembras que en varones.
RECKLINGHAUSEN
Vide: von Recklinghausen (Enfermedad de).
REFSUM (SÍNDROME DE)
El síndrome de Refsum (151) es una heredoataxia polineurítica en probable relación con el metabolismo lípido. El curso es lentamente progresivo, con muerte en unos años. Hay degeneración del sistema nervioso central, ataxia espinocerebelosa, y sordera progresiva. En el aparato ocular lo más llamativo es una retinitis pigmentosa con ceguera nocturna, a lo que se pueden añadir anormalidades pupilares. Alfano y Berger (5) publicaron su asociación con una oftalmoplejia externa congénita.
La herencia es recesiva.
RILEY-DAY (DYSAUTONOMIA FAMILIAR DE)
Este síndrome (152) se limitó inicialmente a reseñar una alacrimia congénita. Más tarde se vio que correspondía a una anomalía genética del sistema nervioso autónomo: trastornos del ritmo cardíaco, hipotensión ortostática, hipertensión arterial paroxística, y labilidad emocional. En el aparato ocular, aparte de la alacrimia, puede haber hipostesia corneal, neuropatía óptica tardía, y ocasionalmente, disfunción oculomotora.
Los genodendros manifiestan una transmisión según el modo autosómico recesivo. El síndrome se debe a una mutación del gen IKBKAP, localizado en el cromosoma 9, banda 9q31. Se da casi siempre en judíos askenazíes.
RUBINSTEIN-TAYBI (SÍNDROME DE)
Se caracteriza este síndrome (154) por braquidactilia, retardo motor y mental, articulaciones hiperextensibles, hiperhirsutismo y hemangiomas. Tres cuartas partes de los afectados tienen estrabismo, más frecuentemente divergente que convergente. La mitad tienen ametropía, y 9 de cada diez manifiestan hendiduras palpebrales antimongoloides y epicanthus.
Es de transmisión autosómica.
SANDHOFF (ENFERMEDAD DE)
La enfermedad de Sandhoff (159,147) se engloba dentro de las gangliosidosis GM2. Es muy parecida a la enfermedad de Tay-Sachs tipo 1 o precoz., por almacenaje y sobrecarga de gangliósidos en el cerebro y otros órganos del cuerpo. La mayoría de los lactantes tienen cara con aspecto de muñeca, macrocefalia, crisis convulsivas, esplenomegalia, y sordera y ceguera progresivas. En fundus oculi es característica la visualización oftalmoscópica de un anillo retiniano perifoveolar blanquecino que contrasta con el color rojo cereza macular. El retraso progresivo de las etapas del desarrollo motor y del lenguaje comienza a los seis meses de vida. Los niños suelen fallecer hacia los tres años de edad.
El gen de la enfermedad se localiza en el brazo largo del cromosoma 5. Se hereda según el modo autosómico recesivo.
SJÖGREN-LARSSON (SÍNDROME DE)
Reúne este síndrome oligofrenia, ictiosis congénita, diplejia espástica, trastornos del habla y temblor de mano (160). En el aparato ocular es frecuente la esoforia, y la esotropia intermitente de ángulo variable. En fondo de ojo puede haber retinitis pigmentosa atípica y lesiones maculares limitadas, con centrotiflosis (disminución de la visión central con conservación de la periférica) moderada.
La herencia es autosómica recesiva.
SMITH-LEMLI-OPITZ (SÍNDROME DE)
Se caracteriza este síndrome (161) por defectos del desarrollo que afectan a muchas partes del cuerpo (cabeza, pulmones, digestivo, riñones, extremidades) en forma de plagiocefalia, crisis convulsivas, hipocolesterolemia, sindactilia o polidactilia, etc. En el aparato ocular es frecuente el epicanthus, agenesia de los puntos lacrimales, estrabismo, y nystagmus. Tiene una elevada mortalidad.
Afecta a 1 de cada 20 ó 30 mil neonatos. La herencia es de tipo autosómico recesivo. La causa es una mutación del gen IVS8-1G-C, localizado en el cromosoma 1q12-q13, que codifica el enzima DHCR7 (7-dehidocolesterol-delta 7 reductasa) (146).
STEELE-RICHARDSON-OLZEWSKI (ENFERMEDAD DE)
El síndrome o enfermedad de Steele-Richardson-Olzewski (15) es una parálisis supranuclear progresiva, que se manifiesta por un síndrome extrapiramidal con parálisis supranuclear y limitación de los desplazamientos verticales de la mirada, produciendo diplopía.
Los más de los casos son esporádicos. Los casos familiares se transmiten de modo autosómico dominante, salvo en una fratría española, en la que la transmisión era autosómica recesiva. El gen responsable es el gen MAPT (Microtubule-Associated Protein Tau), también causante de otras enfermedadesd neurodegenerativas, como el síndrome de Pick, y una variedad de síndrome de Parkinson.
STEINERT (MIOTONÍA DE)
La distrofia miotónica de Steinert (162) es una debilidad de la contracción y un enlentecimiento de la descontracción muscular, que afecta a todos los músculos estriados del cuerpo (cara, músculos oculomotores, manos, faringe, etc.) y a veces a los lisos (digestivos, cardíacos). Se añade calvicie precoz y otras alteraciones cutáneas. En el aparato ocular tiene manifestaciones oculares musculares (blefaroptosis, enlentecimiento de las saccadas, miotonía pupilar) y no musculares (cataratas puntiformes, cataratas polares, anomalías maculares con centrotiflosis). Suele iniciarse en la pubertad o adultez joven.
Se transmite de modo autosómico dominante (179). Perea (139) señala una mayor proporción en varones que en hembras. Los casos que se heredan por la madre son de aparición más precoz, a veces en la lactancia y en la niñez. El gen tarado es una repetición múltiple del triplete de nucleótidos CTG (Citosina-Timina-Guanina), localizada en 19q13.2-q13.3, y codifica una proteinkinasa (145).
STILLING-TÜRK -DUANE (SÍNDROME DE)
Descrito por Stilling (164) (1887), Türk (173) (1899) y Duane (51) (1905), quizás previamente sugerido por Böhm (22) (1845) según interpretación de von Noorden (183), es un síndrome de fibrosis muscular congénita que durante muchos años se creyó que se debía primariamente a fibrosis muscular o a ligamentos anormales, pero hoy se piensa más en un inervación defectuosa, o probablemente supranuclear, y a veces con regeneraciones aberrantes congénitas de los pares craneales III y IV. El síndrome se manifiesta ya al nacer. Consiste en una restricción motora del recto lateral en la que al abducir, la fisura palpebral se abre, y al adducir, se estrecha y el globóculo se retrae (síndrome retractio bulbi). Puede afectar a uno o los dos ojos (61,180). En el 15% de los casos es bilateral, y cuando no es así, el ojo afectado suele ser el izquierdo (cosa que según François (68) es una ley casi general para las afecciones oculares hereditarias). A veces hay heterocromia iridis. Su frecuencia ronda del 1 al 5% de todos los casos de estrabismos. Según Alió et al. (7) es el 1,09% de todos los trastornos oculomotores de la infancia.
La enfermedad suele ser esporádica y no hereditaria, pero en aproximadamente el 20% de los casos es familiar (139), de transmisión autosómica dominante, de penetrancia incompleta. Es más frecuente en mujeres; en la experiencia de Bas Casas et al. (17) con relación mujer/hombre 7/1.
Se han detectado dos genes que pueden producir este síndrome: uno en el brazo largo del cromosoma 8, en 8q13 y otro en el brazo largo del cromosoma 2, en 2q31. También se ha detectado algún caso trisómico con cariotipo 47XX o 46XY/47XY.
Es posible que tenga alguna relación con el síndrome CFEOM-1 y con el síndrome de Okihiro.
STRABISMUS FIXUS
Vide: Fibrosis congénita de los músculos oculomotores (CFEOM) (Síndrome de).
TAY-SACHS (SÍNDROME DE)
Este síndrome (167,157), también conocido como idiocia amaurótica familiar (tipo infantil), es una degeneración de células ganglionares de la retina y cerebro, con gliosis y desmielinización difusa. Está producido por acumulación del gangliósido GM2 en las células ganglionares, lo que concretamente en la oftalmoscopia se detecta como un halo blanquecino que rodea la foveola rosada. Se manifiesta ya en los primeros meses de la vida por retardo mental y disminución de visión. El estrabismo y el nystagmus no son raros (147). Los pacientes suelen fallecer a los 2 ó 3 años de edad. Hay una forma de aparición juvenil de gangliosidosis GM2 en la que no hay ceguera, pero son más intensas las manifestaciones oculomotoras como parálisis supranucleares y oftalmoplegias internucleares. La enfermedad de Sandhoff se considera como la variante 0 de la gangliosidosis GM2. La gangliosidosis GM1 (vide: Gangliosidosis GM1) tiene también múltiples similitudes con la gangliosidosis GM2 o enfermedad de Tay-Sachs.
La enfermedad se transmite generalmente de forma autosómica recesiva, y raramente, dominante. Casi siempre se da en niños judíos askenazíes polacos, y es más frecuente en hembras. El gen causal se localiza en 15q23-q24, y en él se han descrito casi 100 tipos de mutaciones distintas.
TREACHER COLLINS (SÍNDROME DE)
Vide: Franceschetti (Síndrome de).
TRISOMÍA DEL CROMOSOMA 21
Vide: Down (Síndrome de).
TURNER (SÍNDROME DE)
La monogonosomía X o síndrome de Turner (174) es un enanismo hipogenital, con tronco masculino, pterygium colli, cubitus valgus y retardo mental. En el aparato ocular puede haber eso- o exotropía, blefaroptosis, epicanthus, escleróticas azules, glaucoma y despigmentación corio-retiniana. Como estas pacientes sólo tienen un cromosoma X, las discromatopsias protan y deutan son en ellas tan frecuentes como en los varones de la población general.
El cariotipo suele mostrar sólo 45 cromosomas, por cromosoma sexual X0, y en los casos en que no es así, el otro gonosoma es anormal (130). Según Pinto et al. (141) se da en 1 de cada 3.000 nacimientos de hembras.
ULLRICH (SÍNDROME DE)
Este síndrome, descrito en 1951 por Ullrich (176), cuyas diferencias con el síndrome de Bonnevie-Ullrich (175) fueron señaladas por el mismo Ullrich , es una malformación de extremidades (polidactilia, sindactilia, pie zambo), cabeza (hipoplasia mandibular, frente arqueada) y alteraciones genitales (criptorquidia, hipospadias, vagina tabicada). En el aparato ocular puede haber estrabismo, hendiduras palpebrales estrechas, aniridia y glaucoma congénito.
El síndrome se ha visto frecuentemente asociado a la trisomía 13-15. No tiene preferencia según el sexo, y no se ha descrito que se transmita a la descendencia.
Von RECKLINGHAUSEN (ENFERMEDAD DE)
La enfermedad de von Recklinghausen (184) o neurofibromatosis tipo 1 es una alteración neurológica en forma de neurofibromas en múltiples áreas corporales, y alteraciones cutáneas en forma de manchas café con leche. En ojos, es típica la presencia en iris de hamartomas melanocíticos conocidos como nódulos de Lisch. Afecta a una de cada 4.000 personas. Pueyo et al. (150), en una revisión de 44 casos, encontraron una relación niños/niñas de 27/17, y la presencia de estrabismo en 4 casos, de los que 2 casos estaban en relación con la presencia de glioma del nervio óptico.
La enfermedad se debe a un gen localizado en 17q.11.2, responsable de producir la neurofibromina. El 50% de los casos son esporádicos por una mutación del gen (que a partir de entonces puede heredarse), y el otro 50% son heredados de forma autosómica dominante.
WAARDENBURG (SÍNDROME DE)
Vide: Klein-Waardenburg (Síndrome de).
WILDERVANCK (SÍNDROME DE)
Las manifestaciones más características de este síndrome (186) son la parálisis abducens cum retractio bulbi (síndrome de Stilling-Türk-Duane), fusión de vértebras cervicales (anomalía de Klippel-Feil) y sordera congénita de percepción.
En los diferentes genodendros publicados, la afección está casi limitada a las hembras, y sugieren que se transmite de modo dominante ligado al cromosoma X, con letalidad para los varones hemizigotes (130).
WILLIAMS-BEUREN (SÍNDROME DE)
Este síndrome, identificado en 1961 y 1962 (187,20), se caracteriza por estenosis valvular aórtica, dismorfismo facial (cara de diablillo) y retraso mental. Se da en 1 x 20.000 de recién nacidos. Entre las afecciones oculares, Visa et al. (182) encontraron en una revisión de 10 casos, que 6 tenían ametropías (6 miopías, 1 hipermetropía y 2 astigmatismos); y que de esos 6 estrabismos, 5 tenían esotropía y 1 hipertropía. Otras manifestaciones facio-oculares pueden ser sinofridia, anomalías iridianas, y tortuosidad de los vasos retinianos.
La causa está en una deleción en la banda 7q11.23, que incluye entre otros el gen de la proteína elastina. La mayoría de los casos son esporádicos, pero también se ha descrito la transmisión autosómica dominante.
WILSON (SÍNDROME DE)
También conocido como degeneración hepato-lenticular, y como enfermedad de Wilson (188) (1912), se debe a una acumulación de cobre en hígado, riñón, sistema nervioso central y ojos. Se manifiesta lentamente desde la pubertad por cirrosis hepática, trastornos nerviosos (ataxia, temblores) y trastornos oculomotores que suelen empezar por parálisis de los movimientos verticales y se sigue de los horizontales y de la acomodación. En la córnea yuxtalimbal se deposita el anillo de Kayser-Fleischer.
La prevalencia del síndrome es de un caso por 30.000 personas.
La afección se debe a una anomalía del transporte intracelular del cobre. Este transporte está codificado por el gen ATP7B, localizado en el brazo largo del cromosoma 13, en 13q14.3-q21.1. Se transmite del modo recesivo autosómico.
BIBLIOGRAFÍA
Aicardi J, Chevrie JJ, Rousselie F. Le syndrome spasmes en flexion agénesie calleuse, anomalies chorio-rétiniennes. Arch Franç Pédiatr 1969; 26: 1103-1120.
Aicardi J, Barbosa C, Andermann E, et al. Ataxia-ocular motor apraxia: a syndrome mimicking ataxia-telangiectasia. Ann Neurol 1988; 24: 497-502.
Albright F, Burnett CH, Smith PH, Parson W. Pseudo-hypoparathyroidism. An example of Seabright-Bantam syndrome. Endocrinology 1942; 30: 922-932.
Alexander WS.Progressive fibrinoid degeneration of fibrillary astrocytes associated with mental retardation in a hydrocephalic infant. Brain 1949; 72: 373-381.
Alfano JE, Berger JP. Retinitis Pigmentosa. Ophthalmoplegia and Spastic Quadriplegia. Am J. Ophthalmol 1957; 43: 231-240.
Alias Alegre EG, González Viejo MI, Ferrer Novella MC, et al. Síndrome de distiquiasis-linfedema. Acta Estrabol 2006; 35: 151-155.
Alió J, Faci A, Rabinal F. Epidemiología, clínica y pronóstico terapéutico del Síndrome de Duane: Estudio de una muestra de veinticuatro casos clínicos. Acta Estrabológica 1981; 9: 53-68.
Apert E. De lacrocephalosyndactylie. Bull Soc Méd Hôp Paris 1906; 23: 1310-1313.
Arias C, Lanuza A, Ramos F. Agenesia del oblicuo superior asociada a arteria umbilical única y orejas en asa. Acta Estrabológica 1991; 19: 89-92.
Arnalich Montiel F, Rebolleda G, Muñoz-Negrete FJ. SCA-7. Distrofia de conos-bastones en el seno de una ataxia hereditaria. Arch Soc Españ Oftalmol 2005; 80: 679-682.
Arruga A. Nistagmo congénito como primer signo de afecciones neurológicas (a propósito de dos casos de leucodistrofia sudanófila clásica). Acta Estrabológica 1989; 17: 67-70.
Ayuso CM, Baiget F, Palau, V, Volpini V,.Ayuso C. Patología Molecular Hereditaria II. Enfermedades neuromusculares, neurodegenerativas y neurosensoriales. Capítulo 157: 1235-1251. In: Patología Médica. Barcelona: Ed: Farreras Rozman. 2004.
Ayuso CM, Trujillo J, Villaverde C, Vallespín E, Lorda I. Implicaciones genéticas en la Aniridia. Capítulo 3 Sección I: Aspectos básicos en Aniridia. In: Estado Actual y Protocolos de Actuación en la Aniridia Congénita.
Ayuso C. Prevención de las enfermedades oftalmólogicas hereditarias. In: «¿España ve bien? Campaña de Prevención Visual» ONCE y Soc Esp Oftalmología. Capítulo 9: 121-138 (1999).
Balestrazzi P, Baeteman MA, Mattei MG, Mattei JF. Franceschetti syndrome in a child with a de novo balanced translocation (5;13)(q11;p11) and significant decrease of hexosaminidase B. Hum Genet 1983; 64: 305-308.
Bardet G. Sur un syndrome dobésité infantile avec polydactylie et rétinite pigmentaire. Thesis doctoralis. Univ. de París. 1920.
Bas Casas ML, Fernández-Cid C, León Cabello MJ, Montalvo Armas S. Tratamiento quirúrgico en el sdme. de Stilling-Türk-Duane: Nuestra experiencia. Acta Estrabológica 1989; 61-65.
Bassen FE, Kornzweig AL. Malformation of the erythrocytes in a case of atypical retinitis pigmentosa. Blood 1950; 5: 381-388.
Ben Amor M, North P, Collard M, et al. Pierre Marie cerebellar heredo-ataxia. Report of 3 familial cases. Nouv. Presse Méd 1972; 1: 177.
Beuren AJ, Apitz J, Harmjanz D. Supravalvular aortic stenosis in association with mental retardation and a certain facial appearance. Circulation 1962; 26: 1235-1240.
Bloch B. Eigentümliche bisher nicht beschriebene Pigmentaffection. Schweiz med Wochenschr 1926; 56: 404-405.
Böhm L. Das Schielen und der Sehnenschnitt in seinen Wirkungen auf Stellung und Sehkraft der Augen. Berlín.Verlag Dunker und Humblot. 1845
Bojlen, K. Brems T. Hypertelorism (Greig). Acta Path. Microbiol.Scand 1938; 15: 217-258.
Bonnet P, Dechaume J, Blanc E. Lanévrisme cirsoïde de la retine (anévrisme racémeux); ses relations avec lanévrisme cirsoïde de la face et de lanévrisme cirsoïde du cerveau. Bull. Mém Soc Franç Ophtalmol 1938; 51: 521.
Bonnevie K. Erbartzt 1936; 2: 145.
Botelho PJ, Giangiacomo JG. Autosomal-dominant inheritance of congenital superior oblique palsy. Ophthalmology 1996; 103: 1508-1511.
Brachmann W. Ein Fall von symmetrischer Monodactylie durch Ulnadefekt, mit symmetrischer Flughautbildung in den Ellenbeugen sowie anderen Abnormalitäten (Zwerghaftogkeit, Halsrippen, Behaarung). Jahrb Kinderheilk Phys Erzieh 1916; 84: 225-235.
Brice G, Mansour S, Bell R, Collin JR, et al. Analysis of the phenotypic abnormalities in lymphoedema-distichiasis syndrome in 74 patients with FOXc2 mutation or linkage to 16q24. J Med Genet. 2002; 39: 478-83.
Brown S. On hereditary ataxia, with a series of twenty-one cases. Brain 1892; 15: 250.
Cabot J, Boira M, Casale A, García de Oteyza JA. Síndrome de Kearns-Sayre. A propósito de cuatro casos. Acta Estrabológica 1990; 18: 13-18.
Castanera de Molina A. Congenital disorders of ocular motility. Proceedings del 2º International Symposium on Strabismus. Barcelona: Ed. JIMS. 1989.
Castanera de Molina A. Consideraciones sobre la limitación de la abducción en las exotropías congénitas. Acta Estrabológica 1991; 19: 53-61.
Castanera Pueyo. Nistagmus ocular. Madrid: Ed. Paz Montalvo. 1963.
Castiella Acha, JC. Estrabismos acomodativos. Acta Estrabológica 2003; 32; 139-146.
Charcot J. Histologie de la sclérose en plaques. Gaz Hôp (París) 1868; 14: 554-555.
Chediak M. Nouvelle anomalie leucocytaire de caractère constitutionnel et familial. Rev Hémat 1952; 7: 362-367.
Chipont E, López-Correll P, Martínez PA, et al. Un estudio estadístico sobre la incidencia familiar en el estrabismo. Acta Estrabológica 1991; 19: 9-14.
Civantos F. Human chromosomal abnormalities. The Bull., Tulane Univ. Med. Fac. 1961; 20: 241.
Cockayne EA. Dwarfism with retinal atrophy and deafness. Arch Dis Childhood 1936; 11: 1-8.
Cogan DG. Type of congenital ocular motor apraxia presenting jerky head movement. Trans. Am. Acad. Ophthalmol. Oto-laryngol 1952; 36: 433.
Cole DEC, CarpenterTO. Bone fragility, craniosynostosis, ocular proptosis, hydrocephalus, and distinctive facial features: a newly recognized type of osteogenesis imperfecta. J. Pediat. 1987; 110: 76-80.
Cotterman CW, Falls HF. Unilateral developmental anomalies in sisters (status Bonnevie-Ullrich). Am J Hum Genet 1949; 1: 203-213.
Crouzon O. Dysostose cranio-faciale héréditaire. Bull Mém Soc Méd Hôp Paris 1912; 33: 545.
Danlos H. Un cas de cutis laxa avec tumeurs par contusion chronique des coudes et des genoux (xanthome juvénile pseudo-diabetique de MM Hallopeau et Macé de Lépinay). Bull Soc Franç Dermat Syphil 1908; 19: 70-72.
De Lange C. Sur un type nouveau de dégénérescence (typus Amstelodamensis). Arch Méd Enfants 1933; 36: 713-719.
De Morsier G, Gauthier G: La dysplasie olfacto-génitale. Pathol Biol 1963; 11: 1267-1272.
del Río S, Puertas D, Lorenzo Rabanal J. Diagnóstico oftalmológico de la forma infantil de la gangliosidosis GM1.Acta Estrabológica 2000; 29: 61-63.
Doherty EJ, Macy ME, Wang SM, et al. CFEOM3: a new extraocular congenital fibrosis syndrome that maps to 16q24.2-q24.3. Invest Ophthalmol Vis Sci 1999; 40: 1687-1694.
Down JLH. Marriages of consanguinity in relation to degeneration of race. London Hosp Clin Lect & Rep 1866; 3: 224-236.
Drummond KN, Michael AF, Ulstrom RA, Good RA. The blue diaper syndrome: familial hypercalcemia with nephrocalcinosis and indicanuria: a new familial disease with definition of the metabolic abnormality. Am J Med 1964; 37: 928-948.
Duane A. Congenital deficiency of abduction, associated with impairment of abduction, retraction movements, contractions of the palpebral fissure and oblique movements of the eye. Arch Ophthalmol 1905; 34: 133-159.
Duchenne. In: Welander L. Myopathia distalis tarda hereditaria. Acta Med Scand 1951, Suppl 265; 1-124.
Dufier JL, Kaplan J. Oeil et génétique. París: Masson. 2005.
Ehlers E. Cutis laxa. Neigung zu Hömorrhagien in der Haut, Lockerung mehrerer Artikulationen. Dermat Zeitschr 1901; 8: 173-175.
Ellis RWB, van Creveld S. A syndrome characterized by ectodermal dysplasia, polydactily, chondro-dysplasia and congenital morbus cordis. Report of three cases. Arch Dis Childhood 1940; 15: 65.
Engle EC. The molecular basis of the congenital fibrosis syndromes. Strabismus 20002; 10: 125-128.
Esteban de Antonio M. Historia de la estrabología, Hallazgos sorprendentes, curiosos o divertidos, encontrados en nuestras investigaciones. Acta Estrabológica 1993; 21: 103-134.
Estes JW, Morley TJ, Levine IM. A new hereditary acanthocytosis syndrome. Am J Med 1967; 42: 868-881.
Falls HF, Kertesz ED. A new syndrome combining pterygium colli with developmental anomalies of the eyelid and lymphatics of the lower extremities. Trans Am Ophthalmol Soc 1964; 62: 248-275.
Fanconi G, Türler U. Helvet. Paediatri. Acta 1951; 6: 475 (Citado por François 92).
Fernández Muñoz E, Caballero A, Mínguez A. Síndrome de Duane bilateral con distintas malformaciones. Acta Estrabológica 1980; 8: 63-67.
Flaherty MP, Grattan-Smith P, Steinberg A, et al. Congenital fibrosis of the extraocular muscles associated with cortical dysplasia and maldevelopment of the basal ganglia. Ophthalmology 2001; 108: 1313-1322.
Fonseca A. Rev Oftalmol Sao Paulo 1938; 6: 92.
Forsius H, Eriksson AW (1964) Ein neues Augensyndrom mit X-chromosomalen. Myopie, Astigmatismus und Dyschromatopsie, Klin Mbl Augenheilk 1964; 144: 447-457.
Franceschetti A, Zwahlen P. Un syndrome nouveau, la dysostose mandibulo-faciale. Bull schweiz Akad med Wissensch 1944; 1: 60-66.
Franceschetti A, de Morsier G, Klein D. Ueber eine mit Ophthalmoplegia externa progressiva kombinierte infantile Form Arch. J. Klauss-Stift 1945; 20 (suppl): 59.
Franceschetti A, Klein D. Mandibulo-facial dysostosis: New hereditary syndrome. Acta Ophthamol 1949; 27: 143-224.
François J. Lhérédité en ophtalmologie. París: Ed. Masson. 1958. págs 285, etc.
Freeman EA, Sheldon JH. Cranio-carpo-tarsal dystrophy: an undescribed congenital malformation. Arch Dis Childh. 1938; 13: 277-283.
Friedreich N. Ueber degenerative Atrophie der spinalen Hinterstränge. Virchows Arch Pathol Anat Physiol Klin Med 1863; 26: 1-26.
Fuller RW, Weber AN. Hypertelorism in association with sex chromatin abnormality in two siblings. Am J Ment Deficiency 1961-1962; 66: 844-848.
Galán Terraza A, Visa Nasarre J, García Martínez J. Estrabismo en el síndrome de Down. Acta Estrabol 1996; 25: 49-55.
García RE, Friedman WF, Kabach MM, Rowe RD. Idiopathic hypercalcemia and supravalvular aortic stenosis. Documentation of a new syndrome. New Engl J Med 1964; 271: 117-125.
García Lozano I, Jiménez Mateo-Sidrón V, Albrdi P, et a. Nistagmo congénito idiopático. Diagnóstico diferencial. Acta Estrabológica 2003; 33: 37-43.
García Penas J, Puertas Bordallo D, Romero Andújar F, et al. Evolución natural del síndrome oculo-cerebro-renal de Lowe. Acta Estrabol 2006; 35: 39-49.
Gaucher PCE. De lépithélioma primitive de la rate, hypertrophie idiopathique de la rate sans leucémie. Univ. de Paris. Tesis Doctoral 1882.
Geeraets WJ. Ocular syndromes. Filadelfia: Ed. Lea & Febiger. 3ª edición. 1980.
Golden WL, Mamunes P, Kodroff MB. A new familial chondrodystrophy simulating parastremmatic dwarfism. Med. Coll. Va. Quart. 1977; 13(4): 189-191.
Goldenhar M. Associations malformatives de loeil et de loreille, en particulier le syndrome dermoide épibulbaire-appendices auriculaires-fistula auris congenita et ses relations avec la dysostose mandibulo-faciale. J Génét Hum 1952; 1: 243-282.
Goltz RW. Peterson WC, Gorlin RJ, Ravitss HG. Focal dermal hypoplasia. Arch Derm 1962; 86: 708-717.
Gómez de Liaño P, Gómez de Liaño R, Rodríguez Sánchez JM, Gómez de Liaño F. Evoluciones de las exotropias primitivas o permanentes. Acta Estrabológica 1989; 17: 19-24.
Gómez de Liaño R. Alcocer Alfonso P, Ortega Usobiaga J. Oftalmoplejía crónica progresiva externa progresiva «plus». Studium Ophthalmol 2000; 43-45.
Gómez Zaera M, Barrientos A, Arias L, et al Análisis de 6 mutaciones Leber en 31 individuos con atrofia óptica. Estudio de su transmisión en 5 familias. Med Clin (Barcelona) 1999; 112: 326-329.
Gopel W, Barthelmes W. Total vertical ocular paralysis in a case of Friedreich-Nonne-Marie spinocerebellar heredo-ataxia. Z. Aerztl. Fortb. 1970; 64: 1048.
Gorlin RJ, Goltz RW. Multiple nevoid basal cell epithelioma, jaw cysts and bifid rib: A syndrome. New Engl J Med 1960; 262: 908-913.
Greenfield JG. Form of progressive cerebral sclerosis in infants associated with primary degeneration of interfascicular glia. J Neurol Psychopathol 1933; 13: 289-3002.
Greig DM. Hypertelorism. A hitherto undifferentiated congenital craniofacial deformity. Edinburgh Med J 1924; 31: 560-593.
Grouchy J de, Royer P, Salmon C, Lamy M. Délétion partielle des bras longs du chromosome 18. Path. Biol. (Francia) 1964; 12: 579-582.
Gunn RM. Congenital ptosis with peculiar associated movements of the affected lid. Trans Ophthalm Soc UK 1883; 2: 283-287.
Hallermann W. Vogelgesicht und Cataracta congenita. Kl Monatsbl Augenhk 1948; 113: 315.
Hidalgo JM, Medina MC. «Strabismus fixus» congénito. Acta Estrabológica 1984; 12: 55-58.
Higashi O: Congenital gigantism of peroxidase granules. Tohoku J Exp Med 1954; 59: 315-332.
Huntington G. On chorea. Medical and Surgical Reporter 1872; 26: 317-320 y 320-321.
Hurler G. Ueber einen Typ multipler Abartungen, vorwiegend am Skelletsystem. Zeitschr Augenhk 1919; 24: 220.
Izaguirre LB, Playán A, Gonzalvo FJ, et al. Estudio clínico y genético de la neuropatía óptica hereditaria de Leber. Mutaciones en los nucleótidos 11778 y 3460 del ADN mitocondrial. Arch Soc Españ Oftalmol 1998; 73: 527-532.
Jacobs PA, Baikie AG, Court-Brown WM, et al. Evidence for the existence of the human «super female». Lancet 1959; 2: 423-425.
Jarisch . Zur Lehre von den Hautgeschwülsten. Arch Derm Syphil 1894; 28: 163.
Jiang Y, Matsuo T, Fujiwara H, et al.. ARIX gene polymorphisms in patients with congenital superior oblique muscle palsy. Brit J Ophthalmol 2004; 88: 263-267.
Joubert M, Eisenring JJ, Andermann F. Familial dysgenesis of the vermis: a syndrome of hyperventilation, abnormal eye movements and retardation. Neurology 1968; 18: 302-303.
Kearns T, Sayre G. Retinitis pigmentosa, external ophthalmophegia, and complete heart block: unusual syndrome with histologic study in one of two cases. Arch Ophthalmol 1958; 60: 280-289.
King RA, Hearing VJ, Creel DJ, Oetting WS. Albinism. In:Scriver CR, et al. (eds.). Metabolic and molecular bases of inherited diseases. 8ª edición. . Nueva York : McGraw-Hill. 2000.
Kipmanowa I. Neurol Polska 1934; 1617: 348. (Citado por François 92).
Klein D. Albinisme partiel (leucisme) avec surdi-mutité, blépharophimosis et dysplasie myo-ostéo-articulaire. Helvetica Paediatrica Acta, Basel, 1950; 5: 38-58.
Kohn R, Romano PE. Ptosis, blepharophimosis, epicanthus inversus, and telecanthus the syndrome with no name. Am J Ophthalmol 1971; 76: 625-632.
Kremer H, Kuyt LP, van den Helm, et al. Localization of a gene for Mobius syndrome to chromosome 3q by linkage analysis in a Dutch family. Hum Mol Genet 1996; 5: 1367-1371.
Laurence J Z, Moon RC. Four cases of retinitis pigmentosa in the same family and accompanied by general imperfections of development. Ophthal Rev 1866; 2: 32-41.
Lee LV, Pascasio FM, Fuentes FD, Viterbo GH (1976): Torsion dystonia in Panay, Philippines. Adv Neurol 1976; 14: 137-151.
Leigh D. Subacute necrotizing encephalomyelopathy in an infant. J Neurol Neurosurg. Psychiat 1951; 14: 216-221.
Lejeune J, Lafourcade J, Berger R. Trois cas de délétion partielle du bras court dun chromosome 5. Comp. Rend Acad Sci Paris 1963; 257: 3098-3102.
Leri A, Weill J. Phénoméne de Marcus Gunn (synergie palpebro-maxillaire) congénital . Bull. Mém. Soc. Méd. Hôp. Paris 1929; 45: 875-880.
López Areal PL, López Garrido J, Martínez Antón C, et al. Síndrome de Cogan: Apraxia oculomotriz congénita. Acta Estrabológica 1982; 10: 137-142.
Lorenzo Rabanal J, Gallego M, Puertas D, et al. Síndrome de Aicardi adulto. Diagnóstico oftalmológico. Acta Oftalmológica 2001; 31: 13-16.
Louis-Bar D. Sur un syndrome progressif comprenant des télangiectasies capillaires cutanées et conjonctivales symetriques. 1941. In: Louis-Bars syndrome: Ataxia-telangiectasia. a degenerative brain disease. Pediatrics 1958; 21: 526-554.
Lowe CU, Terry M, MacLachlan EA. Organic-aciduria, decreased renal ammonia production, hydrophthalmos and mental retardation. Am J Dis Child 1952; 83: 164-184.
Lowenthal AC, Cummings JF, Wenger DA, et al. Feline sphingolipidosis resembling Niemann-Pick disease type-C. Acta Neuropathologica 1990; 81: 189-197.
Luezas Morcuende JJ, Merino Sanz P, Acero Peña A, et al. Síndrome de Brown: hallazgos histopatológicos y complicaciones quirúrgicas. Acta Estrabol 1997; 26: 21-24.
Magnus W. Norsk Mag. Lägevidensk. H.R. 1889; 13: 265.
Marfan AB. Un cas de deformation congénitale des quatre membres plus prononcées aux extremités characterisée par lallongement des os avec un certain degré damincissement. Bull Mém Soc Med Hôp Paris 1896; 13: 220.
Marie P. Sur lhérédo-ataxie cérébelleuse. Sem. Med. 1893; 13: 444-447.
Marín Amat M. Contribución al estudio de la curabilidad de las parálisis oculares de origen traumatico - substitución funcional de VII por el V par craneal. Oftalmol Hisp-Amer 1918; 18: 70-99.
Marín Amat M. Sur le sindrome ou phénomène de Marcus Gunn. Ann Ocul 1919; 156: 513-528.
Martin JP, Bell J. A pedigree of mental defect showing sex linkage. J Neurol Psychiatry 1943; 6: 154-157.
Merzbacher L. Eine eigenartige familiär-hereditäre Erkrankungsform (aplasia axialis extracorticalis congenita). Zeitschr ges Neurol. Psychiatr 1910; 3: 1-138.
Méténier T. A propos dun cas familial de maladie dEhlers-Danlos; Thesis. Argel. 1939.
Meyer ME. Recueil dOphtalmol 1889; 11: 97.
Michaelides M, Moore AT. The genetics of strabismus. J Med Genet 2004; 41: 641-646.
Möbius PJ. Ueber angeborene doppleseitige Abducens-Facialislähmung. Münch med Wochenschr 1888; 35: 91-94.
Muñoz Negrete FJ, Gómez de Liaño R, Rodríguez Sánchez JM. Our failures in intermittent exotropia surgery. Acta XVII Concilii Europæea Strabologicae Associetatis. Madrid. Ed.J. Murube. 1988. pp. 261-270.
Murube del Castillo J. Desarrollo ontogénico y filogénico del aparato oculomotor extrínseco. Acta Estrabológica 1983; 11: 51-77.
Murube J, Cortés D. Fenotipos oculares ligados al sexo. Ocular sex-linked phenotypes. Barcelona: Ed. Comercial Pujades/Lab. Cusí/AIETI. 1987.
Murube J, Murube E. Near vision accommodation in horizontality with VDT: Why low blinking and dry eye? Advanc Exper Med Biol 2002; 506: 1205-1211.
Naegeli O. Familiärer Chromatophorennävus. Schweiz med Wochenschr 1927; 8: 48.
Nakano N, Yamada K, Fain J, et al.. Homozygous mutations in ARIX (PHOX2A) result in congenital fibrosis of the extraocular muscles type 2. Nat Genet 2001; 29: 315-320.
Navarro Alemany R, Villalonga Gornes PA, Guardia Goma C, Galán Terraza A. Estudio oftalmológico de lños niños con síndrome de Down. Acta Estrabológica 1993; 21: 43-48.
Niemann A. Ein unbekanntes Krankheitsbild. Jahrbuch f Kinderheilkunde 1914; 79: 1-10.
Okihiro MM, Tasaki T, Nakano KK, Bennett BK. Duane syndrome and congenital upper-limb anomalies: a familial occurrence. Arch Neurol 1977; 34: 174-179.
Orssaud C. Strabisme, myopathies, neuro-ophtalmologie. In: Dufier JL, Kaplan J (eds). Oeil et génétique. París: Masson. 2005; 387-435.
Pelizaeus FC. Ueber eine eigenthümliche Form spastischer Lähmung mit Zerebralerscheinungen auf hereditärer Grundlage (multiple sclerose). Arch Psych Nervenkr 1885; 16: 698-710.
Perea García J. Estrabismos. Toledo: Imprenta Artes Gráficas S.A.U. 2008.
Pick L. Niemann-Picks disease and other forms of so-called xanthomatoses. Amer J Med Sci 1933; 185: 601-616.
Pinto C, Valente H, Sphor H, et al. Incidencia de las manifestaciones oculares en el síndrome de Turner (Síndrome de Bonnevie-Ullrich)-. Acta Estrabológica 1993; 21: 9-12.
Prader A, Labhart A, Willi H. Ein Syndrome von Adipositas, Kleinwuchs, Kryptorchismus und Oligophrenie nach Myotonieartigem zustand im Neugeborenenalter. Schweiz Med Wochenschr 1956; 86: 1260-1261.
Prader A, Labhart A, Willi H. Das Syndrom von Imbezilliat, Adipositas Muskelhypotonie, Hypogenitalismus und Diabetes mellitus mit «myatonic»-Anamnese. In: Proceedings of Second International Congress on Mental Retardation. Vienna: S Karger. 19-61.
Prieto Díaz J, Souza Dias, C. Estrabismo. Buenos Aires: Ed. Jims. 2ª edición. 1986.
Puertas Bordallo D, Ruiz-Falcó ML, Abalo JM, et al. Afectación oftalmológica de la distrofia miotónica congénita (Enfermedad de Steinert). Acta Estrabológica 2005; XX: 145-149.
Puertas Bordallo D, Abalo Lojo JM, Ruiz Falco-Rojas ML Alteraciones oftalmológicas en el síndrome de Smith-Lemli-Opitz (SLO) en la infancia. Acta Estrabol 2006; 35: 19-22.
Puertas Bordallo D, de Domingo Barón B, Lozano Vázquez M, et al. Gangliosidosis de tipo infantil (GM 1 tipo1, GM 2 tipo 2). Acta Estrabológica 2006; 35: 23-29.
Puertas Bordallo D, López Conto C, Lozano Vázquez M, et al. Displasia óculo-aurículo-vertebral o síndrome de Goldenhar. Acta Estrabol 2006; 35: 127-130.
Puertas Bordallo D, López Couto C, Ruiz Falcó-Rojas ML, et al. Manifestaciones neuro-oftalmológicas en la enfermedad de Leber. Acta Estrabol 206; 35: 131-133.
Pueyo V, González I, Ferrer C, et al. Manifestaciones oculares de la neurofibromatosis tipo I en la infancia. Acta Estrabológica 2005; 2: 71-75.
Refsum S. Heredopathia atactica polyneuritiformis. Oslo: Ed. Grundt Tanum. 1946.
Riley CM, Day RL, Greeley DM, Langford WS. Central autonomic dysfunction with defective lacrimation;. Report of 5 cases. Pediatrics 1949; 3: 468-478.
Rodríguez Sánchez JM, Cores González J, Rebolleda Fernández G, Mendez Llatas M. Aplasia congénita del oblicuo superior. Acta Estrabológica 1989; 17: 71-74.
Rubinstein JH, Taybi H. Broad thumbs and toes and facial abnormalities. Am J Dis Child 1963; 105: 588-608.
Ruiz Maldonado R, Carnevale A, Tamayo L, de Montiel EM. Focal dermal hypoplasia. Clinical Genetics 1974; 6: 36-45.
Saá JA, Valls I, Puertas D, et al. Síndrome de Prader-Willi: Hallazgos oftalmológicos en tres casos. Acta Estrabológica 2000; 29: 57-60.
Sachs B. On arrested cerebral development with special reference to its cortical pathology. J Nerv Mental Dis 1887; 14: 541-553.
Sánchez Ramos C, Romero Martín M, Domínguez Carmona M. Frecuencia de heteroforias en universitarios usuarios de ordenador. Acta Estrabol 1994; 22: 71-76.
Sandhoff K, van Echten G. Ganglioside metabolism. Topology and regulation Adv Lipid Res 1993; 26: 119-138.
Sjögren T, Larsson T. Oligophrenia in combination with congenital ichthyosis and spastic disorders; a clinical and genetic study. Acta Psych Neurol Scand. 1957. Suppl 113.
Smith DW, Lemli L, Opitz JM. A newly recognized syndrome of multiple congenital anomalies. J Pediatr 1964; 64: 210-217.
Steinert H. Ueber das klinische und anatomische Bild des Muskelschwunds der Myotoniker. Deutsch Zeitschr Nervenheilk 1909; 37: 58-104.
Stevenson AC, Davidson BC. Genetic counselling. Londres: Heinemann, 2ª ed. 1976.
Stilling J. In: Untersuchungen über die Entstehung der Kurzsichtigheit. Wiesbaden, J.F. Bergmann, 1887: 13.
Streiff EB. Dysmorphie mandibulo-faciale (tête doiseau) et altérations oculaires. Ophthalmologica 1950; 120: 79-83.
Sulzberger MB. Ueber eine bisher nicht beschriebene congenitale Pigmentanomalie (incontinentia pigmenti). Arch Derm Syph (Berlin) 1927-8; 154: 19-32.
Tay W. Symmetrical changes in the region of the yellow spot in each.eye of an infant. Trans Ophthalmol Soc UK 1881; 1: 55-57.
Temtamy SA. Genetic factors in hand malformations. Thesis. The Johns Hopkins University. Baltimore. 1966.
Temtamy SA, Shoukry AS, Ghali I, et al. The Duane radial dysplasia syndrome: an autosomal dominant disorder. Birth Defects Orig Art Ser 1975; 11: 344-345.
Thouvenin D, Arné JL. Parálisis unilateral congénita del III par y malformaciones de las extremidades ¿síndrome similar al de Möebius? Acta Estrabol 1995; 24: 31-38.
Tnacheri OI, Ibrahimy W, Benharbit M, et al. Syndrome de Crouzon associé à une atteinte oculaire, à une malformation Chiari type I et à une syringomyélie. J Franç Ophtalmol 2009; 32(1S): 181.
Toro Solá MA, Kistenmacher ML, Punnett HH, di George AM. Focal dermal hypoplasia. Syndrome in a male. Clinical Genetics 1975; 7: 325-327.
Türk S: Bemerkungen zu einem Falle von Retraction des Auges. Zbl. Pract. Augenheilk. 1899; 23: 14-18.
Turner HH. Syndrome of infantilism, congenital webbed neck and cubitus valgus. Endocrinology 1938; 23: 566-574.
Ullrich O. Ueber typische Kombinationsbilder multipler Abartungen. Zeitschr. f. Kinderhk. 1930; 49: 271-276.
Ullrich O. Der Status Bonnevie-Ullrich in Rahmen anderer Dyscranio-Dysphalangien. Ergebn inn Med Kinderh 1951; 2: 412-453.
van Vliet AGM, Waardenburg PG, Forsius H, Eriksson AW. Nystagmographical studies in Aland eye disease. Acta Ophthalmol 1973; 51: 782-790.
Vasella F. Lütschg J, Mumenthaler M. Cogans congenital oculomotor apraxia in two successive generations. Dev. Med. Child. Neurol 1972; 14: 788-796.
Vico Ruiz E, Calvo González C, Gómez de Liaño R. Patología de la motilidad ocular en la distrofia miotónica de Steinert Acta Estrabológica, 2005; 34: 3-9.
Vieira L, GarcíaI, Martíns R, Barreto V. Síndrome de Duane. Algunos aspectos clínicos. Acta Estrabológica 1988; 16: 25-31.
Villalonga Gornes PA, Guardia Goma C, Navarro Alemany R, Galán Terraza A. Síndrome de fibrosis generaslizada de los músculos extraoculares. Acta Estrabológica 1993; 21: 3-8.
Visa Nasarre J, Gracia Martínez J, Núñez Pérez X. Manifestaciones oculares en el síndrome de Williams. Acta Estrabológica 2005; 34: 31-34.
von Noorden GK. The history of strabismology. Oostende: Ed. JP Wayenborgh. 2002. The history of European strabismology. p. 77.
von Recklinghausen F. Ueber die multiplen Fibrome der Haut und ihre Beziehung zu den multiplen Neuromen. Berlin: Editor August Hirschwald. 1882.
Waardenburg PJ. A new syndrome combining development anomalies of the eylelids, eyebrows and nose root with pigmentary defects of the iris and head hair and with congenital deafness. Dystopia canthi medialis et punctorum lacrimalium lateroversa, hyperlasia supercilii nedialis et radicis nasi, heterochromia iridium totalis sive partialis, albinismus circumscriptus (surdimutitas). Amer J Hum Genetics 1951, 3: 195-253.
Wildervanck LS. Een cervico-oculo-acusticus syndroom. Nederl. T. Geneesk. 1960; 104: 2600-2605.
Williams JCP, Barrat-Boyes BG, Lowe JB. Supravalvular aortic stenosis. Circulation 1961; 24: 1311-1318.
Wilson SAK. Progressive lenticular degeneration: a familial nervous disease associated with cirrhosis of the liver. Brain 1912; 34: 295-507.
Windmiller J. Whalley PJ, Fink CW. Cockaynes syndrome with chromosomal analysis. Am J. Dis Child. 1963; 105: 204-208.
Wyburn-Mason R. Arteriovenous aneurysm of mid-brain and retina, facial naevi and mental changes. Brain 1943; 66: 163-203.