SEMINARIO DE CASOS CLÍNICOS
Hipertrofia de epitelio pigmentario de la retina en el síndrome oral-facial-digital tipo 1
GUTIÉRREZ MONTERO O1, RECHE-SAINZ JA1, STOICA B1
Hospital Universitario de Fuenlabrada.
Madrid. España.
1 Licenciado en Medicina.
RESUMEN
Introducción:
El síndrome oral-facial-digital tipo I es un trastorno malformativo que afecta principalmente a cara, cavidad oral y dedos, y que además puede cursar con poliquistosis renal y con alteraciones del sistema nervioso central. No se habían descrito alteraciones oculares asociadas.Caso clínico:
Dos pacientes hermanas gemelas de 17 años de edad afectadas por el síndrome oral-facial-digital tipo I acudieron para ser valoradas oftalmológicamente. En las dos pacientes la agudeza visual era de la unidad decimal sin corrección en ambos ojos, y la exploración del segmento anterior normal. En el fondo de ojo de ambas pacientes, se objetivaban múltiples lesiones hiperpigmentadas bilaterales que se localizaban en áreas extensas de la periferia retiniana de ambos ojos, compatibles con hipertrofia del epitelio pigmentario con morfología de «huella de oso». Dado la edad de las pacientes y el carácter bilateral y extenso de las lesiones, se les realizó una exploración colonoscópica que resultó ser negativa al no hallarse pólipos adenomatosos.Palabras clave:
síndrome oral-facial-digital tipo I, hipertrofia de epitelio pigmentario, poliposis colónica.INTRODUCCIÓN
El síndrome oral-facial-digital tipo I es un trastorno del desarrollo caracterizado por malformaciones de la cara, cavidad oral y dedos. Se transmite con una herencia dominante ligada al cromosoma X. Su expresividad clínica es muy variable, y en algunos casos también se observan malformaciones del sistema nervioso central y poliquistosis renal. A continuación se describen las manifestaciones sistémicas y oculares de dos hermanas gemelas afectadas por este síndrome.
CASO CLÍNICO
Dos hermanas gemelas univitelinas de 17 años de edad (A y B) con diagnóstico de síndrome oral-facial-digital tipo 1, nos fueron referidas para revisión oftalmológica. Fenotípicamente ambas presentaban hipertelorismo, puente nasal ancho, hipoplasia de las alas nasales, philtrum del labio superior alargado y pabellones auriculares de implantación baja (fig. 1). Además tenían paladar hendido, con frenillo bucal y malposición dentaria. En las manos se les observó braquidactilia. Respecto al sistema nervioso se les detectó agenesia del cuerpo calloso, además de un retraso mental más acusado en la paciente A. Esta misma paciente A manifestaba una hemiparesia braquiocrural derecha con discreta espasticidad. Otro hallazgo común a ambas fue la poliquistosis renal. Se les realizó el estudio del gen OFD1 asociado a este síndrome, situado en el brazo corto del cromosoma, pero no se identificó ninguna mutación.
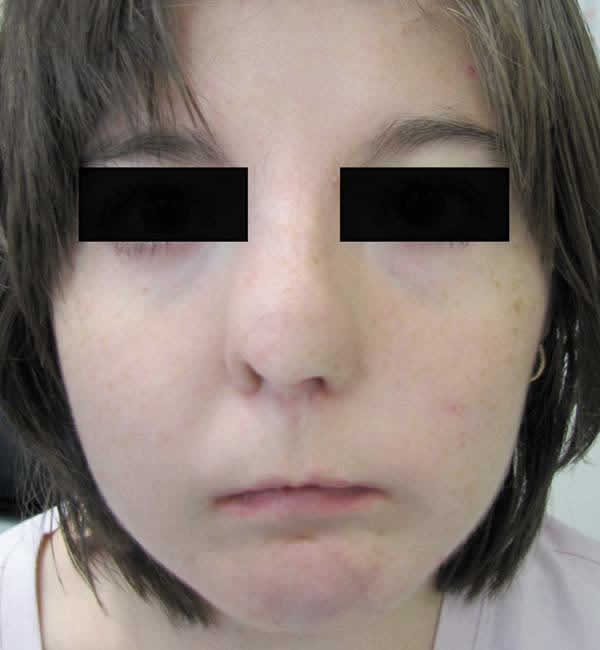
Fig. 1: Aspecto facial
de la paciente A: hipertelorismo, puente nasal ancho, hipoplasia de alas nasales
y philthum largo.
En la exploración oftalmológica, la agudeza visual espontánea era de la unidad decimal en ambos ojos de ambas pacientes. En la refracción ciclopléjica manifestaron una hipermetropía de +1 dioptría. La exploración del polo anterior fue anodina. En el fondo de ojo, la mácula y papila presentaban un aspecto normal, pero en áreas extensas de la periferia temporal superior e inferior de ambos ojos (figs. 2 y 3), se objetivaban múltiples lesiones hiperpigmentadas, de aspecto plano, de formas redondeadas y ovales, con tamaños variables que oscilaba entre 0,5 a 1,5 diámetros papilares, y que además se presentaban en acúmulos. Dichas lesiones eran compatibles con hipertrofia del epitelio pigmentario con morfología de «huella de oso».
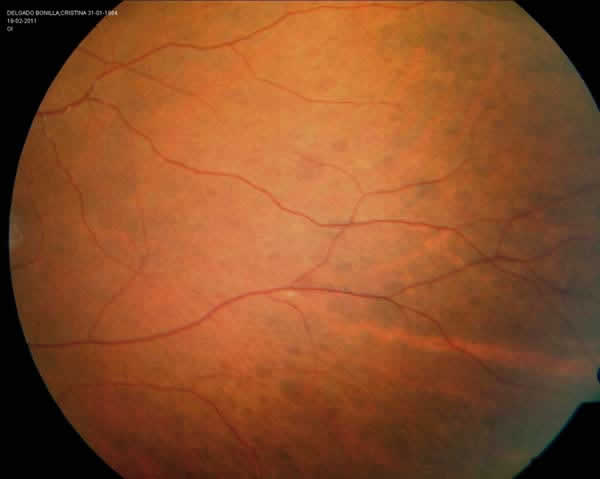
Fig. 2: Retina temporal
periférica del OI de la paciente A.
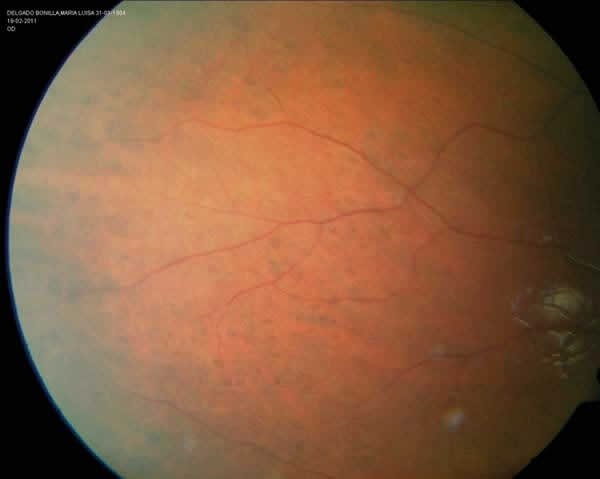
Fig. 3: Retina temporal
periférica del OD de la paciente B.
DISCUSIÓN
El síndrome oral-facial-digital tipo 1 (OFD1) fue primeramente descrito en 1954 por Papillon-Léage-Psaume (1). Su incidencia es de 1 entre 50.000 recién nacidos vivos (2). La dismorfia facial es la característica principal del síndrome (1-3) e incluye hipertelorismo, puente nasal ancho, hipoplasia de las alas nasales, frenillo bucal, paladar hendido (80%) (1,3) y lengua bífida o lobulada (30%) (3). Las anomalías dentarias incluyen hipondotia y malposición. Las alteraciones digitales son frecuentes (50-70%) (1,3) y comprenden braquilactilia, sindactilia, clinodactilia y, más raramente, polidactilia. La poliquistosis renal es habitual (44%) (2). Las malformaciones del sistema nervioso son variables (40%) (1-3), e incluyen agenesia del cuerpo calloso, quistes aracnoideos, trastornos cerebelosos, hidrocefalia y pueden asociar retraso mental. En la Literatura revisada, no ha sido descrita ninguna alteración ocular específica. La variabilidad clínica de este síndrome es muy alta, y el diagnóstico puede ser difícil en sus formas clínicas menores (2). Aproximadamente el 75-80% de los casos son esporádicos en los casos hereditarios. La herencia está ligada al sexo y es letal en los varones (1-3). El gen OFD1 localizado en el cromosoma X, se expresa de forma ubicua en los tejidos adultos. La variabilidad clínica del síndrome incluso entre sujetos de una misma familia podría deberse a los diferentes grados de mosaicismo somático resultante de la inactivación del cromosoma X (1). Se han descrito hasta 29 mutaciones genéticas posibles del gen OFD1 (2).
La hipertrofia del epitelio pigmentario de la retina (HEPR) es una lesión pigmentada de tipo benigno. Existen formas típicas que pueden ser solitarias o múltiples, y formas atípicas (4). Las lesiones típicas suelen ser planas, grises o negruzcas, redondeadas u ovaladas con márgenes discretos y cuando son múltiples se organizan en acúmulos que simulan «huellas de oso». Las lesiones atípicas son ovales o fusiformes, múltiples pero más separadas y muchas veces se observa cómo uno de los márgenes de las lesiones está despigmentado.
El diagnóstico diferencial se ha de establecer con la hiperplasia reactiva del epitelio pigmentario de la retina secundaria a traumas o inflamaciones, hamartomas combinados de retina, melanomas metastásicos y síndromes paraneoplásicos con proliferación melanocítica uveal difusa y bilateral (4).
Las lesiones atípicas, multifocales, bilaterales de la HEPR se asocian con varios síndromes de poliposis colónica (4,5). La poliposis adenomatosa familiar (PAF) es un trastorno autosómico dominante que se caracteriza por la existencia de múltiples pólipos en el colon y recto que de no ser tratados, se malignizan en la práctica totalidad de los casos (4,5). El gen responsable llamado APC, ha sido identificado en el cromosoma 5 (5q21-q22) (4). El síndrome de Gardner asocia además, osteomas y tumores de piel y tejidos blandos, y el síndrome de Turcot, tumores del sistema nervioso central (meduloblastomas) (4).
Las lesiones atípicas de HEPR están presentes aproximadamente en dos tercios de los casos de PAF (4,5). Al ser un trastorno de herencia autosómica dominante es obligada la exploración funduscópica de los familiares en busca de lesiones, pero su ausencia no excluye el diagnóstico de PAF (4,5).
Dado el carácter bilateral y extenso de las lesiones y la edad de nuestras pacientes, se le realizó una exploración colonoscópica que resultó ser negativa. En los padres no se observaron lesiones HEPR en el fondo de ojo y por lo que se desechó realizar un estudio genético específico.
En los casos presentados del síndrome oral-facial-digital tipo I, las lesiones de HEPR fueron un hallazgo funduscópico casual, y no hubo evidencia de que se asociasen a PAF.
BIBLIOGRAFÍA
- Ferrante MI, Giorgio G, Feather SA, Bulfone A, Wright V, Ghiani M et al. Identification of the gene for oral-facial-digital type 1 syndrome. Am J Hum Genet 2001; 68: 569-570.
- Thauvin-Robinet C, Cossée M, Comier-Daire V, Van Maldergem L, Toutain A, Alembik Y et al. Clinical, molecular, and genotype-phenotype correlation studies from 25 cases of oral-facial-digital syndrome type 1: a French and Belgian collaborative study. J Med Genet 2006; 43: 54-61.
- Feather SA, Woolf AS, Donnai D, Mlacolm S, Winter RM. The oral-facial-digital syndrome type 1 (OFD1), a cause of polycystic kidney disease and associated malformations, maps to Xp22.2-Xp22.3. Human Molecular Genetics 1997; 6: 1163-1167.