 Fig. 1
Fig. 1Dres. Pueyo M1, Ferrer E1, Torrón C1, González I1, Leache JJ2, Honrubia FM3
Hospital Miguel Servet. Zaragoza. España.
(1) Servicio de Oftalmología. Doctor en Medicina y Cirugía.
(2) Servicio de Otorrinolaringología. Licenciado en Medicina y Cirugía.
(3) Servicio de Oftalmología. Profesor Titular.
Introducción
En 1945, Cogan (1) describió la asociación de queratitis intersticial (QI) de etiología no luética y una disfunción vestíbulo-auditiva (DVA) consistente en vértigo, acúfenos y pérdida de audición. Aunque su etiología y patogenia son desconocidas, la sugerencia de Oliner y col (2) en 1953 de que esta entidad pueda ser el reflejo de una enfermedad sistémica, ha sido mantenida en posteriores publicaciones (3-5).
Haynes y col (6), en 1980 establecieron los siguientes criterios para su diagnóstico y clasificación. El síndrome de Cogan (SC) típico presenta una DVA consistente en un síndrome vertiginoso similar al Menière, con acúfenos y pérdida de audición progresiva que puede llegar a la sordera total, pudiendo aparecer antes o después de los síntomas visuales, que se caracterizan por una QI no sifilítica, habitualmente bilateral y con tendencia recidivante, pudiéndose asociar a iritis y hemorragias conjuntivales o subconjuntivales. El SC atípico se caracteriza por la presencia de otra lesión inflamatoria ocular significativa, asociada o no a QI; escleritis, epiescleritis, oclusión de la arteria retiniana, coroiditis, hemorragias retinianas, edema de papila y exoftalmus; así como los casos de iritis, conjuntivitis, hemorragia conjuntival o subconjuntival sin asociarse a QI, pero con DVA. Si la DVA no es similar al síndrome de Menière, o aparece más de dos años después de la sintomatología ocular, se considera a su vez una forma atípica.
Presentamos el caso de un paciente con síndrome de Cogan atípico, con mala evolución de la afectación auditiva, a pesar del tratamiento, y con buena respuesta de su lesión oftalmológica.
Caso clínico
Mujer de 23 años, sin antecedentes patológicos de interés, que acude a urgencias por presentar hipoacusia bilateral progresiva de 15 días de evolución, en el oído derecho y de tres días en el oído izquierdo, acompañada de acúfenos bilaterales y vértigo rotatorio con sensación nauseosa. La exploración otoscópica era normal, con rinne positivo y weber indiferente presentando nistagmus bilateral, siendo el resto de la exploración neurológica normal.
La audiometría tonal mostró una hipoacusia perceptiva bilateral importante. Tanto la Rx de tórax como los estudios hematológicos fueron normales, incluyendo la ECA y la VSG. Tras el ingreso se realizó un TAC craneal y de ambos oídos, siendo informado como normal; igualmente resultaron negativos tanto la serología luética, como el estudio inmunitario (incluyendo la determinación de autoanticuerpos). Se inició tratamiento con corticoides orales (30 mg deflazacort/día), minodipino y complejo vitamínico B, evolucionando en dos semanas a una mejoría del síndrome vertiginoso, persistiendo la hipoacusia bilateral, que empeoró progresivamente.
A los dos meses presentó enrojecimiento, fotofobia, dolor y disminución de la agudeza visual (AV), por lo que fue remitida a nuestro servicio, con la siguiente exploración oftalmológica:
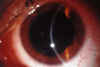
La AV con su mejor corrección era de 6/10 en ojo derecho (OD) y de 4/10 en ojo izquierdo (OI). El examen biomicroscópico revelaba una queratitis intersticial bilateral, más importante en el OI, acompañada de una moderada iritis en el OI (figs. 1 y 2). El resto de la exploración oftalmológica fue normal. La paciente se diagnosticó de síndrome de Cogan típico, y se instauró tratamiento con midriáticos y corticoides tópicos. Ante este diagnóstico se vuelve a repetir el estudio autoinmune, que sigue sin alteraciones y se abandona el tratamiento con corticoides orales.
Fig. 1
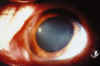 Fig. 2
Fig. 2
Durante los dos últimos años, la paciente ha presentado episodios repetitivos de queratitis e iritis, uni o bilaterales, que han remitido siempre al tratamiento tópico. Además, la paciente ha presentado un episodio de proptosis de OI de una semana de duración, coincidiendo con un cuadro de malestar general, astenia y sudoración, que cedió al tratamiento con corticoides orales y tópicos. En la actualidad la paciente se encuentra con una grave alteración auditiva y una exploración oftalmológica normal sin tratamiento; y realizándose controles periódicos por parte de internistas en previsión de posibles alteraciones generales.
Discusión
Diferentes etiologías han sido postuladas como causa del SC, incluyendo la autoinmune, la hereditaria o la infecciosa, pero el mecanismo definitivo no ha sido bien aclarado. Se han observado con frecuencia signos de síntomas respiratorios previos, que sugirieron una etiología infecciosa (7,8), sin embargo, no se ha encontrado una asociación con un agente infeccioso específico, vacuna o toxina (8). Últimamente, el origen autoinmune es defendido por varios autores, apoyándose en diferentes hallazgos: la presencia de autoanticuerpos frente a tejidos de oído interno y córnea (9,10); la observación de infiltrado linfocitario y de células plasmáticas en la córnea junto con mediadores de inmunidad celular (9); la presencia de varios casos de incremento de linfocitos circulantes T y B, con signos de consumo de complemento (9); y la buena respuesta al tratamiento inmunosupresor (7-9,11).
La incidencia del SC es mínima, habiéndose descrito sólo unos 140 casos en la literatura médica, siendo la edad media de presentación de esta rara enfermedad entorno a los 25 años (2-63 años) y la distribución por sexos similar (7,8). Los primeros signos de la enfermedad pueden ser oculares o auditivos, sin predominio significativo de uno de ellos (12). El intervalo entre las manifestaciones oftalmológicas y las auditivas varia desde cero (simultáneas) a más de un año (13,14).
El cuadro puede iniciarse con síntomas inespecíficos, sobre todo en el síndrome atípico, consistentes en pérdida de peso, fiebre, cefaleas, siendo muy frecuente en algunas series la presencia (40%) de artralgias y mialgias (8). La disfunción vestibuloauditiva (DVA) se caracteriza por un comienzo brusco con náuseas, vómito, vértigo rotatorio e hipoacusia neurosensorial lentamente progresiva, muchas veces bilateral y que afecta a todas las frecuencias.
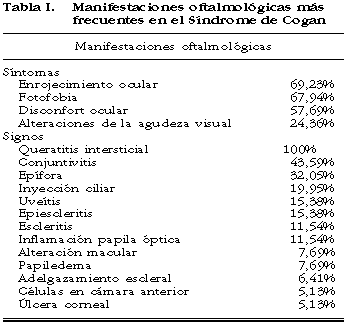
Analizando los datos de la revisión más extensa publicada (78 casos) (8), se recogen los siguientes datos de afectación oftalmológica (tabla I). El enrojecimiento, fotofobia, disconfor ocular y alteraciones en la agudeza visual han sido síntomas muy frecuentes. Las alteraciones oculares son a menudo fugaces, y a veces son necesarios exámenes repetitivos para demostrar la queratitis intersticial (8). La manifestación más usual es la queratitis intersticial con infiltrado granular de distribución moteada situado predominantemente en la mitad posterior de la córnea (14,15). El examen oftalmológico frecuentemente revela conjuntivitis y epífora. La inyección ciliar, uveítis, epiescleritis, escleritis, la inflamación de la papila óptica sugestiva de neuritis óptica, alteraciones maculares, papiledema, adelgazamiento escleral, células en cámara anterior y ulceración corneal, son signos menos frecuentes. También han sido descritos: oclusión de la vena central de la retina, blefaritis, ptosis, proptosis, uveítis "granulomatosa", hemorragia vítrea, nódulos conjuntivales y glaucoma (8). La AV fluctúa en el curso de la enfermedad y con el tratamiento. Se han presentado cegueras eventuales en aproximadamente un 5% de los ojos (8).
La afectación sistémica es más frecuente en el SC atípico y lo más frecuente es la presencia de artralgias (40%), fiebre (30%), cefalea (20%) y dolor abdominal (15%) (8). En un 30% de los casos se asocia a una vasculitis diseminada con afectación de piel, músculo, ganglios linfáticos, hueso, riñón, tubo digestivo y tejido sinovial (7,8). En el síndrome típico la vasculitis diseminada es infrecuente y se presenta una forma localizada en forma de aortitis en el 10-15% de los casos, que puede llegar a producir una insuficiencia aórtica y requerir reemplazamiento valvular (9,16).
También existe la posibilidad de afectación del sistema nervioso central, con perineuritis y meningismo, habiéndose recogido un caso de SC atípico con microinfartos cerebrales que respondió satisfactoriamente al tratamiento inmunosupresor (17). Analíticamente suele haber una leucocitosis discreta con VSG elevada, en numerosas ocasiones superior a 100, siendo relativamente frecuente la presencia de anemia normocítica y normocrómica (7,8).
El diagnóstico es fundamentalmente clínico, debiendo descartarse una sífilis congénita, una tuberculosis, infecciones víricas, una oncocercosis y una sarcoidosis, que producen una queratitis intersticial no ulcerativa y estromal, similar a la descrita en el SC, así como las vasculitis necrotizantes sistémicas, periarteritis nodosa, artritis reumatoidea, granulomatosis de Wegener y arteritis de Takayasu, que pueden presentarse con afectación ocular y DVA (7,8). Ademas deben tenerse en cuenta Menières bilaterales, la sífilis congénita, la rubeola congénita y la laberintitis viral como posibles causas de una DVA.
La afectación ocular del SC es usualmente autolimitada, y el tratamiento tópico con corticoides y midriáticos basta para controlar la sintomatología (7-9,13,14,18). La sintomatología vertiginosa y los acúfenos suelen desaparecer lentamente, pero la hipoacusia evoluciona hacia la sordera permanente intensa o total en el 95% de los no tratados con corticoides (6). En cuanto a la evolución de los tratados los resultados son muy dispares, y mientras unos recogen buenos resultados en todos los pacientes tratados dentro de las cuatro primeras semanas (7,19); otros, entre los que incluimos nuestro caso, recogen sorderas permanentes en pacientes correctamente tratados (20). Este tratamiento consiste en prednisolona (o equivalente) a dosis de 1-2 mg/kg/día fraccionados en 4 tomas, disminuyendo la dosis tras dos semanas, pasando a régimen alterno durante 4 semanas, para suprimirla de 3 a 6 meses más tarde (7,8,16,17). Si tras las dos primeras semanas no se evidencia mejoría, se recomienda reducir rápidamente la dosis hasta suprimirla (7,8). En casos de vasculitis con gran extensión o en casos particularmente graves, se han usado agentes inmunosupresores (ciclofosfamida, ciclosporina y azathiopina) con buenos resultados (8,11,17).
Ante la presencia de una queratitis intersticial u otras lesiones oculares inflamatorias, junto a una disfunción vestibuloauditiva, ha de plantearse el diagnóstico de esta rara enfermedad. La oposición entre la relativa benignidad de los signos oculares y la gravedad de la afectación auditiva, teniendo en cuenta la posibilidad de que la enfermedad debute con manifestaciones oftalmológicas, ha de hacernos pensar en el diagnóstico de un SC, a fin de instituir una corticoterapia a dosis eficaces desde la aparición de los primeros signos auditivos. Es por esto importante un mejor conocimiento de este infrecuente síndrome por parte de los oftalmólogos.
Resumen
El síndrome de Cogan (SC) es una rara enfermedad caracterizada por la asociación de queratitis intersticial (QI) aguda no sifilítica junto a episodios agudos de vértigo, acúfenos y pérdida auditiva. La sordera permanente, de moderada a intensa, es la consecuencia más grave de la disfunción vestibuloauditiva (DVA) y se da en el 95% de los pacientes no tratados. Las lesiones oculares responden satisfactoriamente a la terapia con corticoides, lo cual se opone al carácter severo y frecuentemente definitivo de la afectación auditiva.
Presentamos el caso clínico de una paciente afecta de SC atípico, que provocó sordera intensa, y hacemos énfasis en el diagnóstico y tratamiento.
Palabras clave
Síndrome Cogan, queratitis intersticial, disfunción vestibuloauditiva, síndromes oculo-auditivos.
Summary
Cogan's Syndrome (SC) is an uncommon disease characterized by the association of acute non-syphilitic interstitial keratitis (QI) associated to acute episodes of vertigo, tinnitus, and hearing loss. Moderate to severe permanent deafness is the most serious sequela of the vestibular-auditory dysfunction (DVA) and is found in 95% of non-treated patiens. Ocular involvement responds well to corticosteroids therapy but hearing loss is severe and often irreversible.
We present the case of one patient with atypical SC, with DVA causing severe deafness. We emphasize the diagnosis and treatment.
Key words
Cogan's Syndrome, intersticial keratitis, vestibular-auditory dysfunction, oculo-auditive syndromes.
Bibliografía